
Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus Strains
2.2. Primary Adult Murine Neuronal Cultures
2.3. Establishment of Latent Infection
2.4. Reactivation with Stress Hormones
2.5. Reactivation with Receptor Agonists & Antagonists
2.6. Reactivation with Protein Inhibitors
2.7. Quantitation of HSV Viral Load
2.8. Plaque Assay
2.9. Quantitation of HSV Gene Expression
2.10. Quantification and Statistical Analysis
3. Results
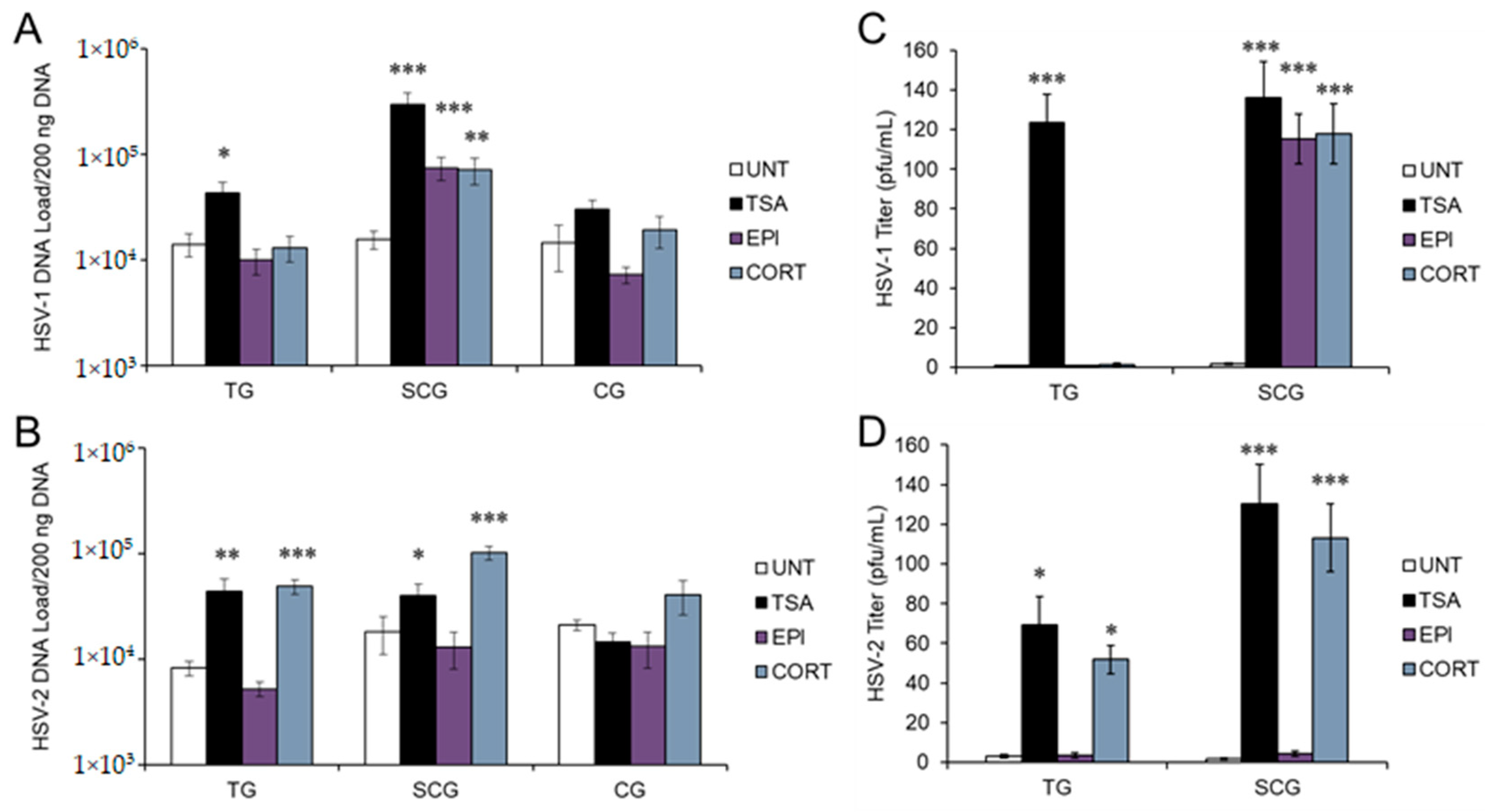
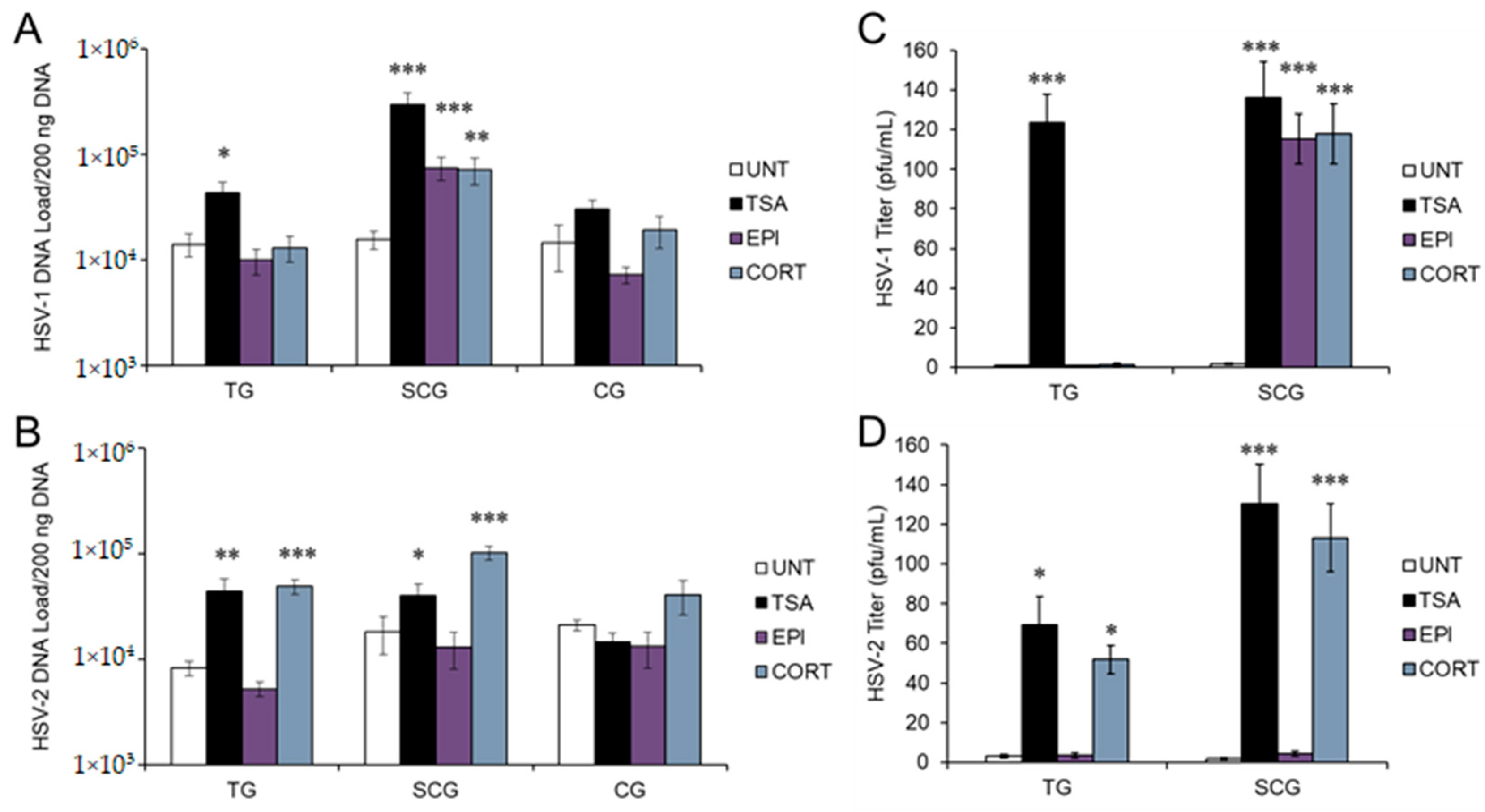
3.1. Stress Hormones Differentially Induce HSV Reactivation In Vitro
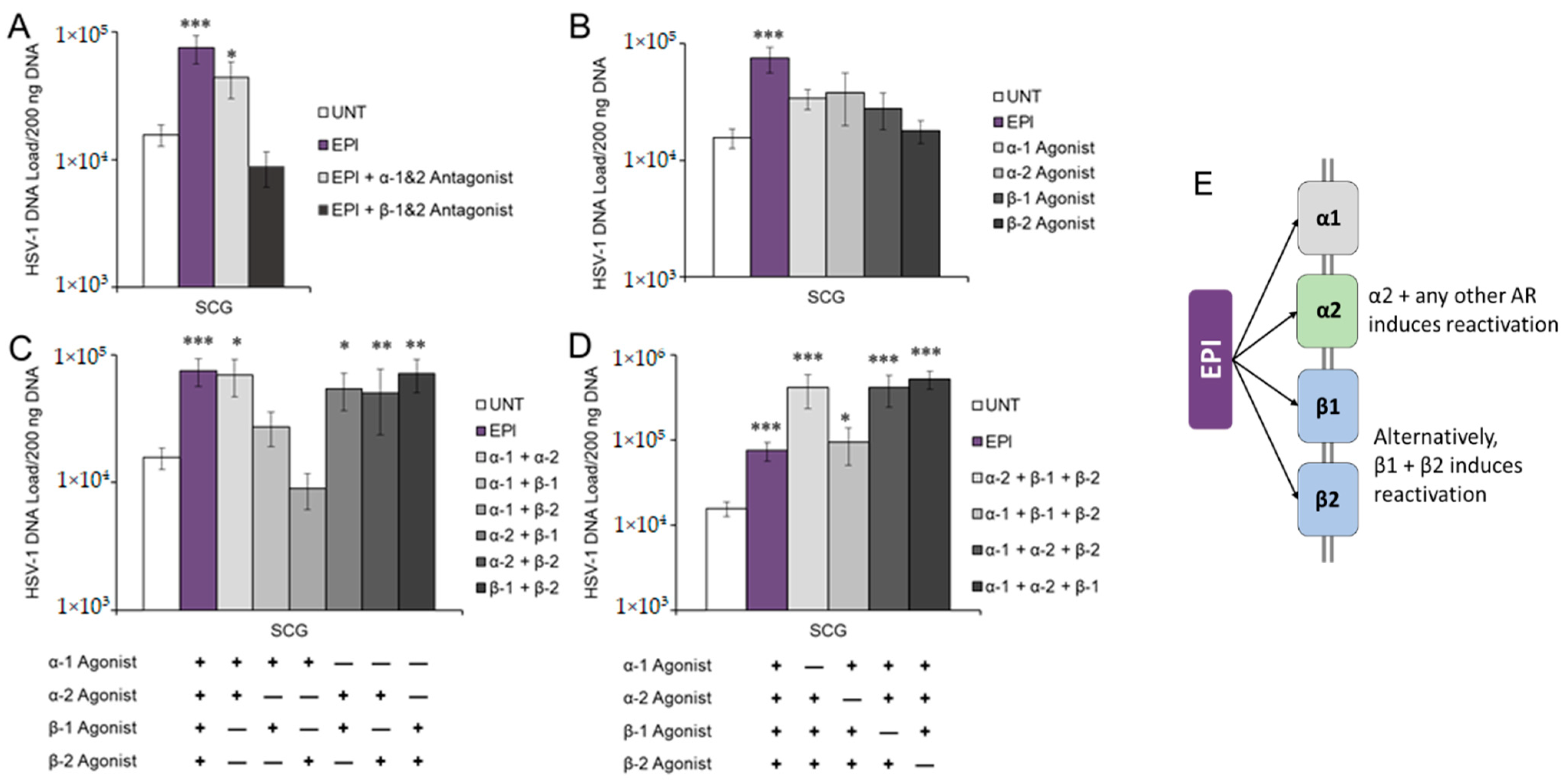
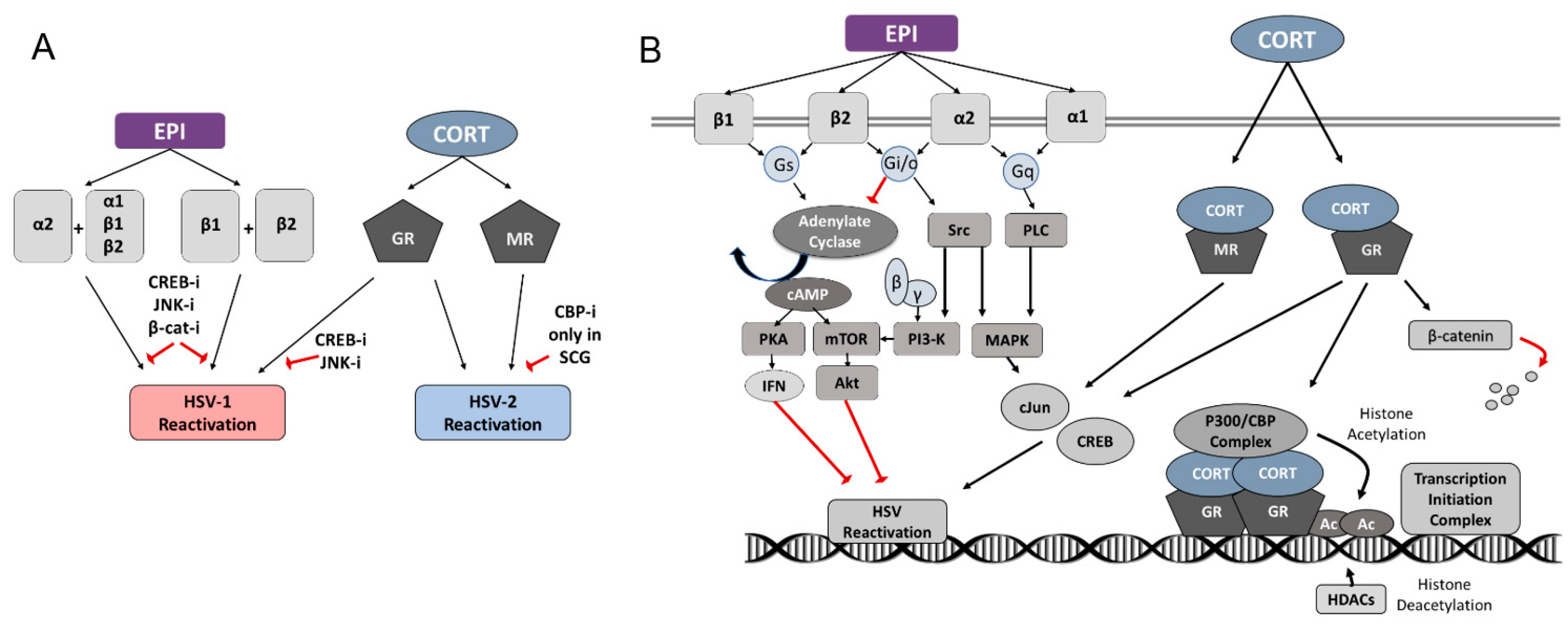
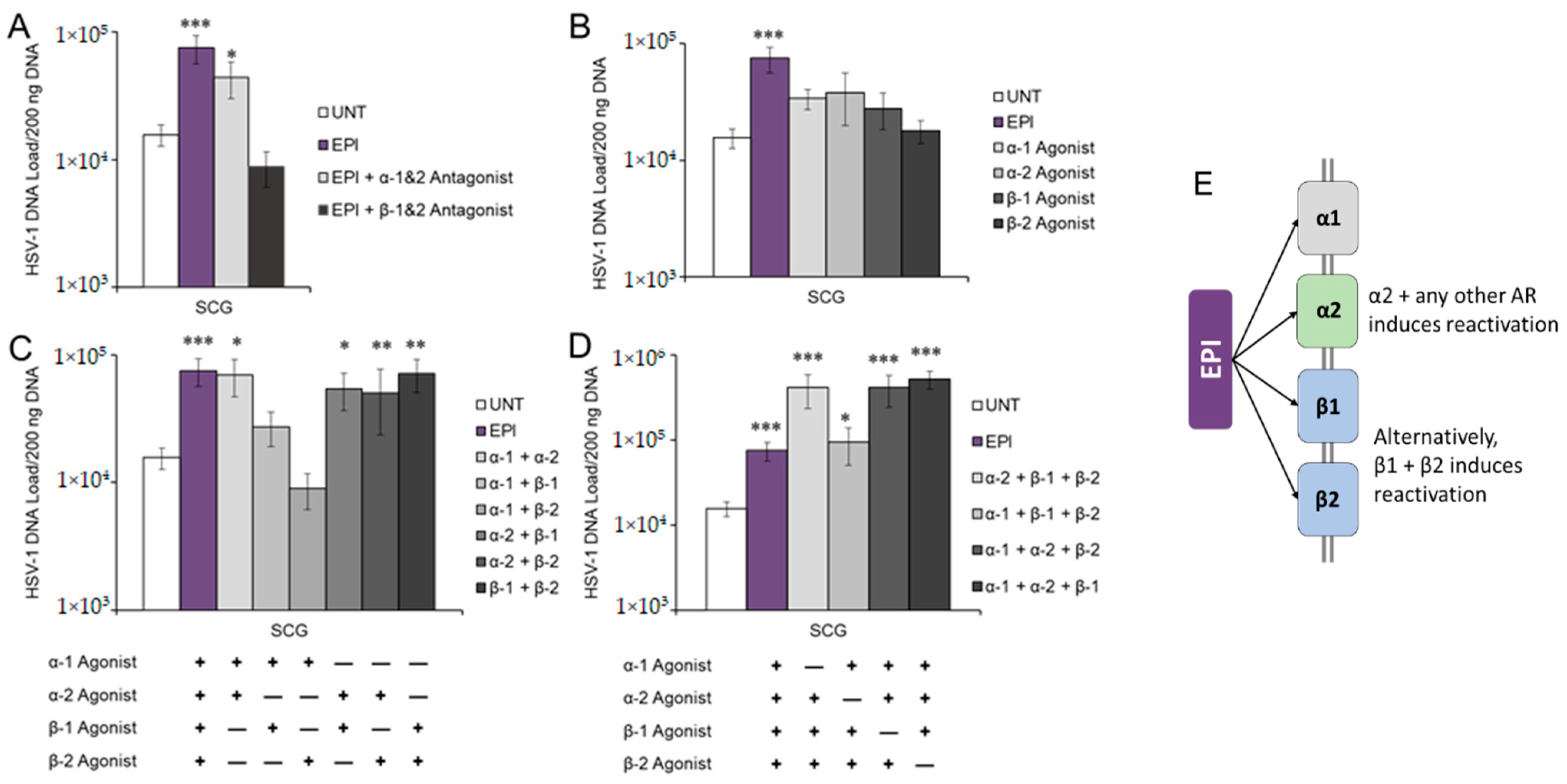
3.2. Adrenergic Receptor Specificity of EPI-Induced Reactivation of HSV-1
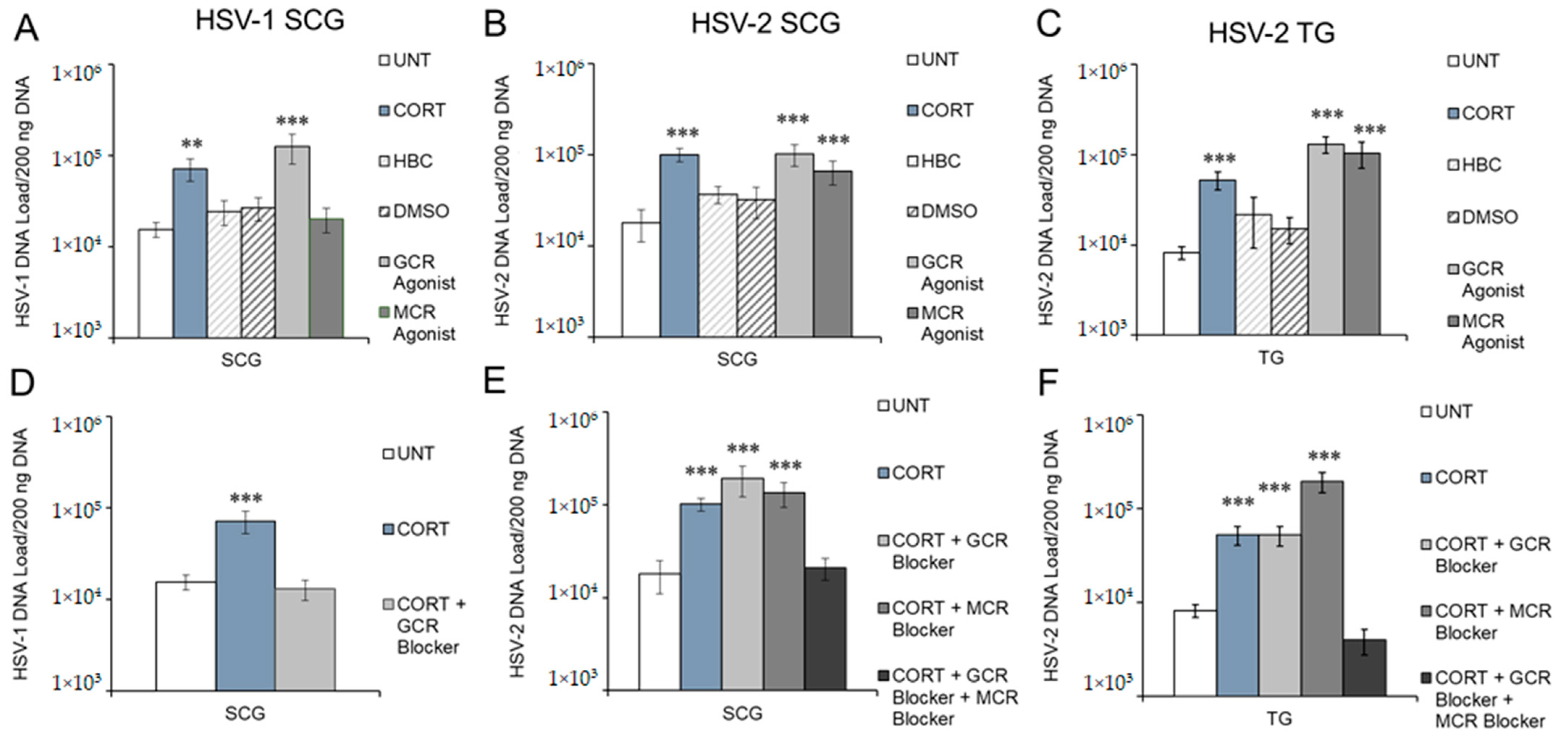
3.3. Corticosteroid Receptor Specificity of CORT-Induced Reactivation of HSV-1 and HSV-2
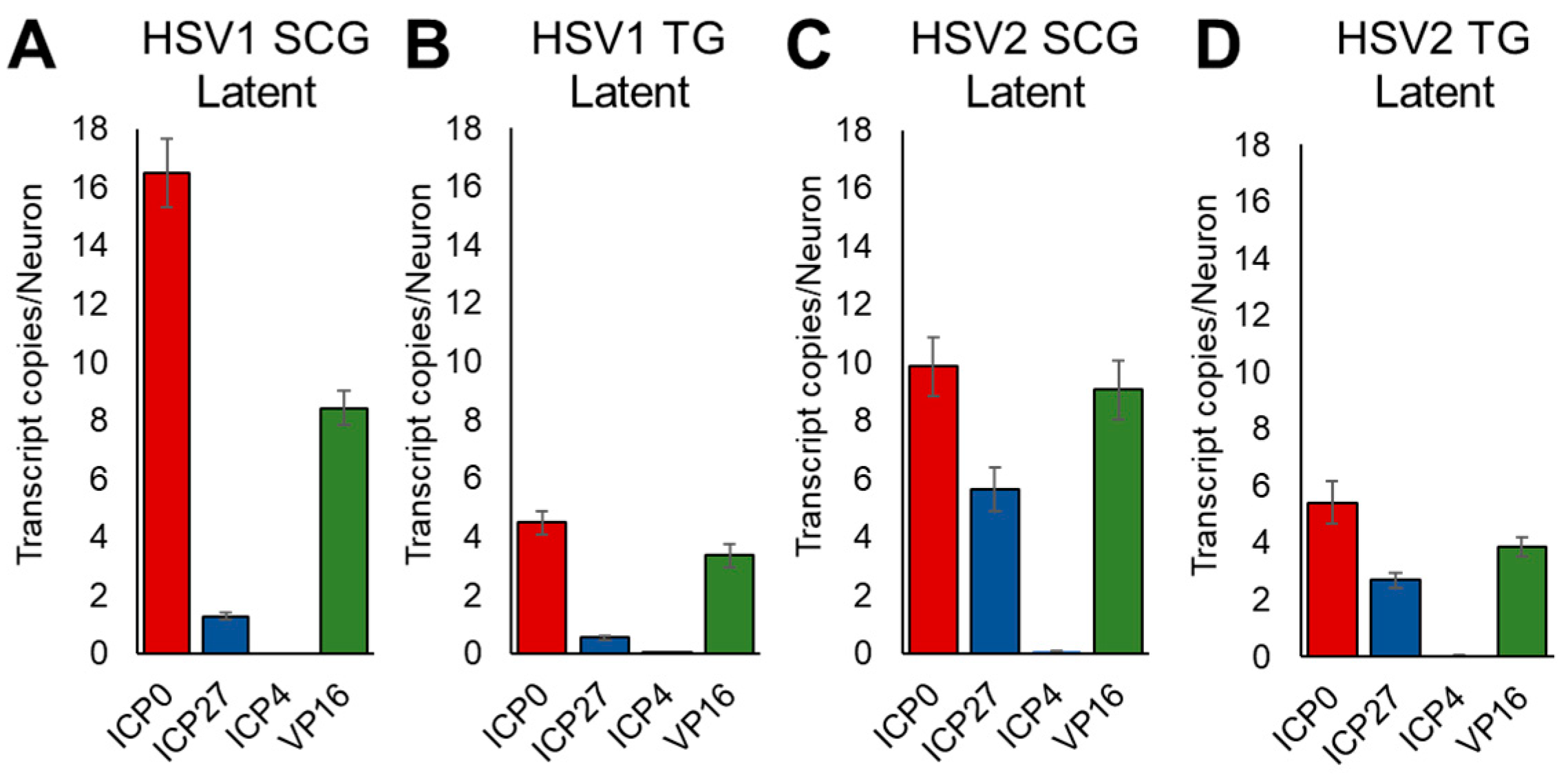
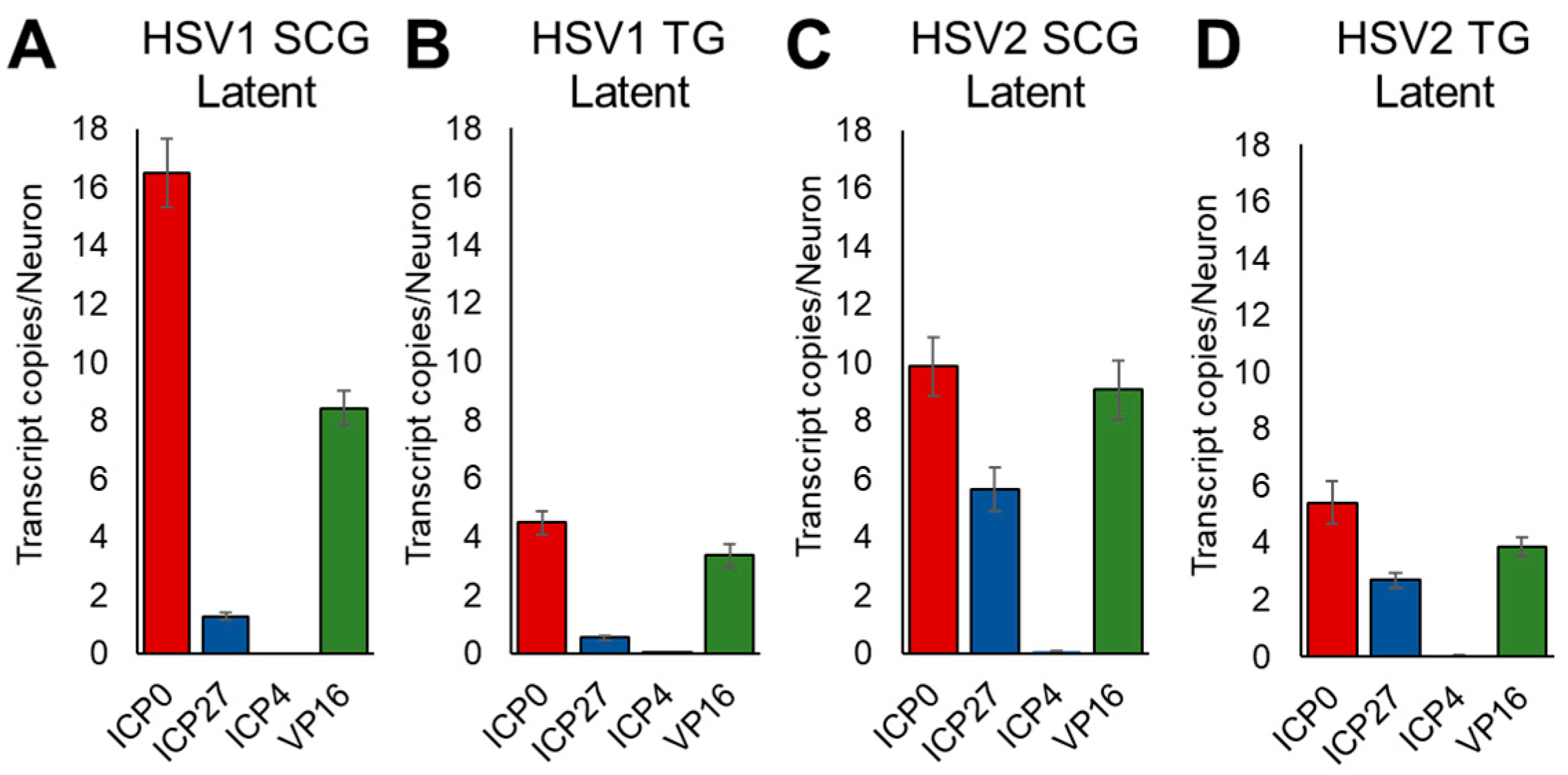
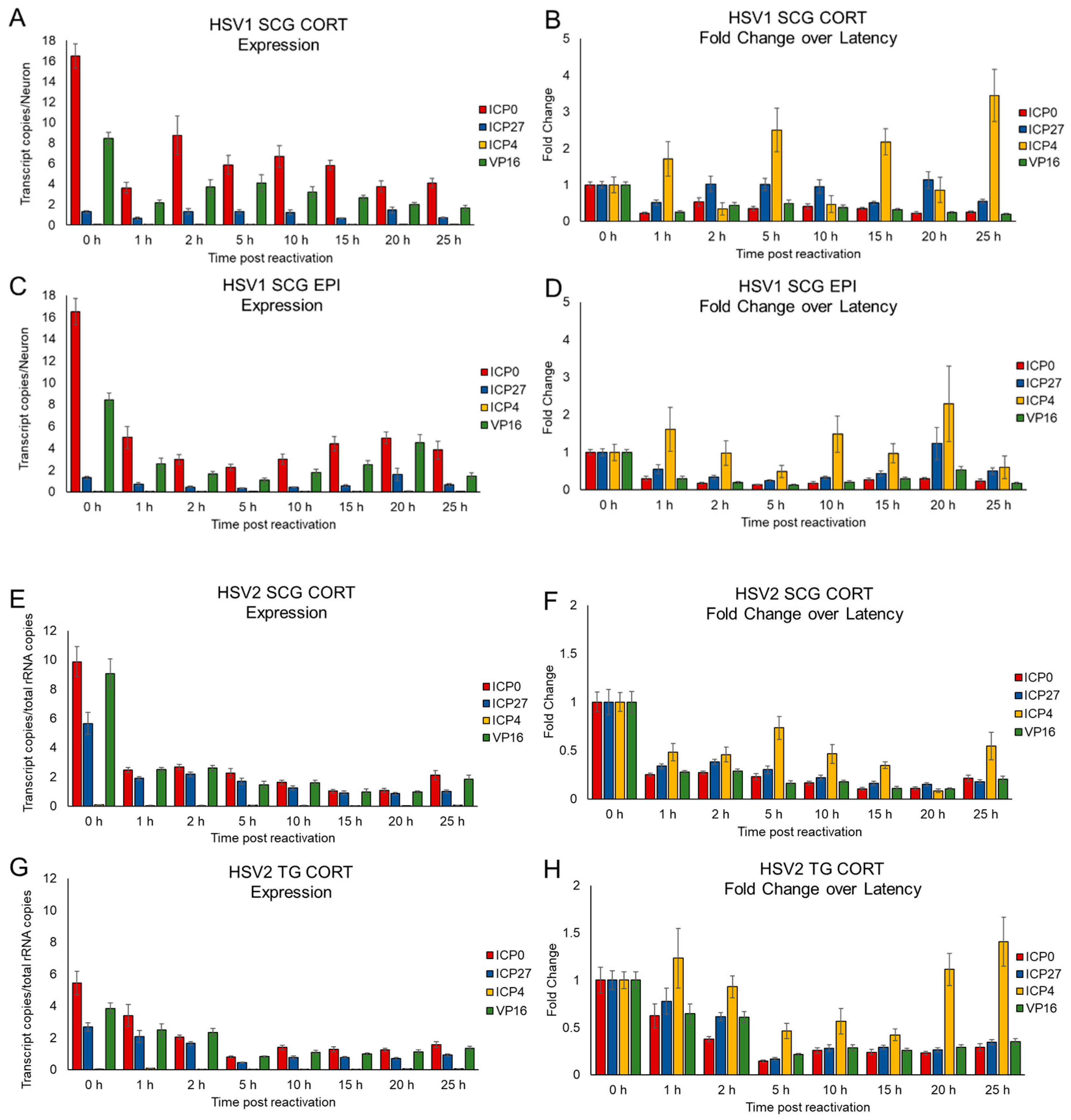
3.4. Kinetics of HSV Gene Expression Differ after EPI and CORT Treatment, but Also Depend on Neuron Type
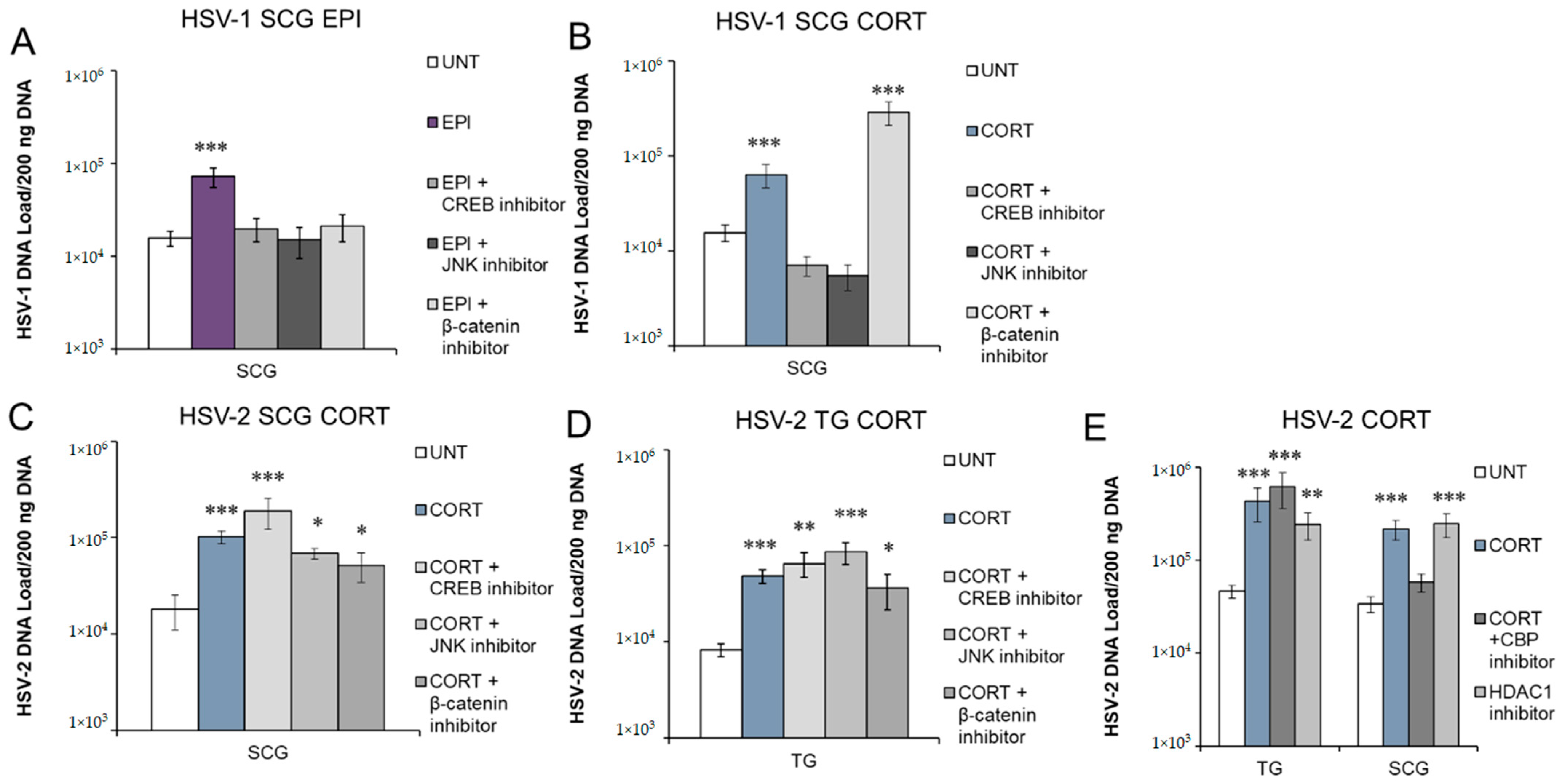
3.5. Inhibition of Protein Kinases and Transcription Factors Selectively Block Stress Hormone-Induced Reactivation of HSV-1 and HSV-2
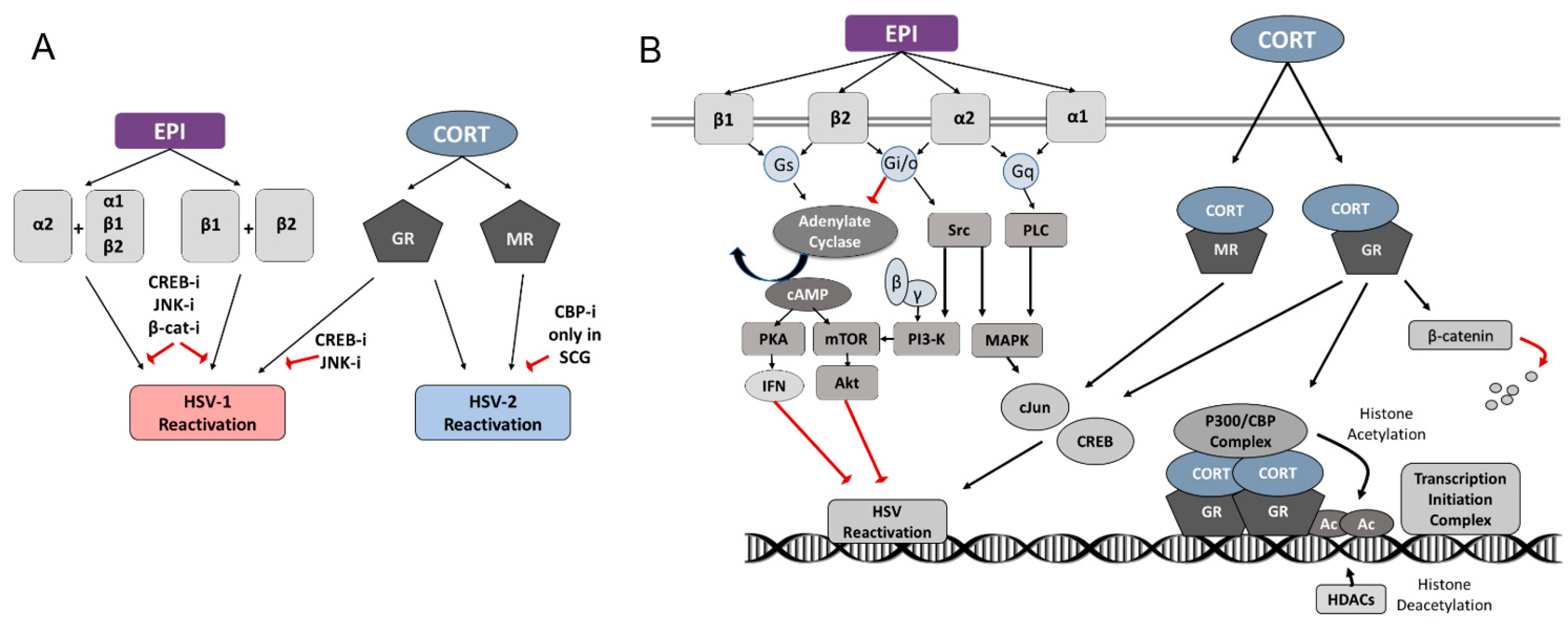
4. Discussion
4.1. HSV-1
4.2. HSV-2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benedetti, J.; Corey, L.; Ashley, R. Recurrence rates in genital herpes after symptomatic first-episode infection. Ann. Intern. Med. 1994, 121, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, W.E.; Coombs, R.W.; Benedetti, J.; Critchlow, C.; Corey, L. Recurrences after oral and genital herpes simplex virus infection. Influence of site of infection and viral type. N. Engl. J. Med. 1987, 316, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Tronstein, E.; Johnston, C.; Huang, M.L.; Selke, S.; Magaret, A.; Warren, T.; Corey, L.; Wald, A. Genital shedding of herpes simplex virus among symptomatic and asymptomatic persons with HSV-2 infection. J. Am. Med. Assoc. 2011, 305, 1441–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, L.; Adams, H.G.; Brown, Z.A.; Holmes, K.K. Genital herpes simplex virus infections: Clinical manifestations, course, and complications. Ann. Intern. Med. 1983, 98, 958–972. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Gilden, D.H. The expanding spectrum of herpesvirus infections of the nervous system. Brain Pathol. 2001, 11, 440–451. [Google Scholar] [CrossRef]
- Picard, F.J.; Dekaban, G.A.; Silva, J.; Rice, G.P. Mollaret’s meningitis associated with herpes simplex type 2 infection. Neurology 1993, 43, 1722–1727. [Google Scholar] [CrossRef]
- Delahunt, J.; Mellsop, G. Hormone changes in stress. Stress Med. 1987, 3, 123–134. [Google Scholar] [CrossRef]
- Logan, H.; Lutgendorf, S.K.; Hartwig, A.; Lilly, J.; Berberich, S.L. Immune, stress, and mood markers related to recurrent oral herpes outbreaks. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1998, 86, 48–54. [Google Scholar] [CrossRef]
- Cohen, F.; Kemeny, M.E.; Kearney, K.A.; Zegans, L.S.; Neuhaus, J.M.; Conant, M.A. Persistent stress as a predictor of genital herpes recurrence. Arch. Intern. Med. 1999, 159, 2430–2436. [Google Scholar] [CrossRef] [Green Version]
- Chida, Y.; Mao, X. Does psychosocial stress predict symptomatic herpes simplex virus recurrence? A meta-analytic investigation on prospective studies. Brain Behav. Immun. 2009, 23, 917–925. [Google Scholar] [CrossRef]
- Kwon, B.S.; Gangarosa, L.P.; Burch, K.D.; deBack, J.; Hill, J.M. Induction of ocular herpes simplex virus shedding by iontophoresis of epinephrine into rabbit cornea. Investig. Ophthalmol. Vis. Sci. 1981, 21, 442–449. [Google Scholar]
- Willey, D.E.; Trousdale, M.D.; Nesburn, A.B. Reactivation of murine latent HSV infection by epinephrine iontophoresis. Investig. Ophthalmol. Vis. Sci. 1984, 25, 945–950. [Google Scholar]
- Padgett, D.A.; Sheridan, J.F.; Dorne, J.; Berntson, G.G.; Candelora, J.; Glaser, R. Social stress and the reactivation of latent herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 1998, 95, 7231–7235. [Google Scholar] [CrossRef] [Green Version]
- Gold, M.S.; Dastmalchi, S.; Levine, J.D. Alpha2-adrenergic receptor subtypes in rat dorsal root and superior cervical ganglion neurons. Pain 1997, 69, 179–190. [Google Scholar] [CrossRef]
- DeLeon, M.; Covenas, R.; Chadi, G.; Narvaez, J.A.; Fuxe, K.; Cintra, A. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res. 1994, 636, 338–342. [Google Scholar] [CrossRef]
- Ives, A.M.; Bertke, A.S. Stress Hormones Epinephrine and Corticosterone Selectively Modulate Herpes Simplex Virus 1 (HSV-1) and HSV-2 Productive Infections in Adult Sympathetic, but Not Sensory, Neurons. J. Virol. 2017, 91, e00582-17. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.M.; Sedarati, F.; Javier, R.T.; Wagner, E.K.; Stevens, J.G. Herpes simplex virus latent phase transcription facilities in vivo reactivation. Virology 1990, 174, 117–125. [Google Scholar] [CrossRef]
- Hill, J.; Patel, A.; Bhattacharjee, P.; Krause, P. An HSV-1 chimeric containing HSV-2 latency associated transcript (LAT) sequences has significantly reduced adrenergic reactivation in the rabbit eye model. Curr. Eye Res. 2003, 26, 219–224. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Hill, J.M.; Stanberry, L.R.; Bourne, N.; Kurawadwala, J.F.; Krause, P.R. The characteristic site-specific reactivation phenotypes of HSV-1 and HSV-2 depend upon the latency-associated transcript region. J. Exp. Med. 1996, 184, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B.; Loutsch, J.M.; Marquart, M.E.; Hill, J.M. Stress-associated immunomodulation and herpes simplex virus infections. Med. Hypotheses 2001, 56, 348–356. [Google Scholar] [CrossRef]
- Zahwa, H.; Yorty, J.L.; Bonneau, R.H. Elevated maternal corticosterone during lactation hinders the neonatal adaptive immune response to herpes simplex virus (HSV) infection. Brain Behav. Immun. 2008, 22, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Sinani, D.; Cordes, E.; Workman, A.; Thunuguntia, P.; Jones, C. Stress-induced cellular transcription factors expressed in trigeminal ganglionic neurons stimulate the herpes simplex virus type 1 (HSV-1) infected cell protein 0 (ICP0) promoter. J. Virol. 2013, 87, 13042–13047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, K.S.; Zhu, L.; Thunuguntla, P.; Jones, C. Antagonizing the Glucocorticoid Recept or Impairs Explant-Induced Reactivation in Mice Latently Infected with Herpes Simplex Virus 1. J. Virol. 2019, 93, e00418–e00419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostler, J.B.; Harrison, K.S.; Schroeder, K.; Thunuguntla, P.; Jones, C. The Glucocorticoid Receptor (GR) Stimulates Herpes Simplex Virus 1 Productive Infection, in Part Because the Infected Cell Protein 0 (ICP0) Promoter Is Cooperatively Transactivated by the GR and Kruppel-Like Transcription Factor 15. J. Virol. 2019, 93, e02063-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostler, J.B.; Jones, C. Stress Induced Transcription Factors Transactivate the Herpes Simplex Virus 1 Infected Cell Protein 27 (ICP27) Transcriptional Enhancer. Viruses 2021, 13, 2296. [Google Scholar] [CrossRef] [PubMed]
- Ostler, J.B.; Thunuguntla, P.; Hendrickson, B.Y.; Jones, C. Transactivation of Herpes Simplex Virus 1 (HSV-1) Infected Cell Protein 4 Enhancer by Glucocorticoid Receptor and Stress-Induced Transcription Factors Requires Overlapping Kruppel-Like Transcription Factor 4/Sp1 Binding Sites. J. Virol. 2021, 95, e01776-20. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.X.; Chen, J.X.; Young, C.S.; Silverstein, S. Reactivation of latent herpes simplex virus by adenovirus recombinants encoding mutant IE-0 gene products. J. Virol. 1990, 64, 4489–4498. [Google Scholar] [CrossRef] [Green Version]
- Halford, W.P.; Schaffer, P.A. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 2001, 75, 3240–3249. [Google Scholar] [CrossRef] [Green Version]
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 2001, 75, 6143–6153. [Google Scholar] [CrossRef] [Green Version]
- Bertke, A.S.; Swanson, S.M.; Chen, J.; Imai, Y.; Kinchington, P.R.; Margolis, T.P. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 2011, 85, 6669–6677. [Google Scholar] [CrossRef] [Green Version]
- Bertke, A.S.; Ma, A.; Margolis, M.S.; Margolis, T.P. Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J. Virol. 2013, 87, 6512–6516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanez, A.A.; Harrell, T.; Sriranganathan, H.J.; Ives, A.M.; Bertke, A.S. Neurotrophic Factors NGF, GDNF and NTN Selectively Modulate HSV1 and HSV2 Lytic Infection and Reactivation in Primary Adult Sensory and Autonomic Neurons. Pathogens 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Wilson, A.C.; Chao, M.V.; Mohr, I. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev. 2012, 26, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Ferguson, S.S.G.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.-T.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β-Arrestin-Dependent Formation of β2 Adrenergic Receptor-Src Protein Kinase Complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Chabre, O.; Conklin, B.R.; Brandon, S.; Bourne, H.R.; Limbird, L.E. Coupling of the alpha 2A-adrenergic receptor to multiple G-proteins. A simple approach for estimating receptor-G-protein coupling efficiency in a transient expression system. J. Biol. Chem. 1994, 269, 5730–5734. [Google Scholar] [CrossRef]
- Ramirez, M.T.; Sah, V.P.; Zhao, X.L.; Hunter, J.J.; Chien, K.R.; Brown, J.H. The MEKK-JNK Pathway Is Stimulated by α1-Adrenergic Receptor and Ras Activation and Is Associated with in Vitroand in Vivo Cardiac Hypertrophy. J. Biol. Chem. 1997, 272, 14057–14061. [Google Scholar] [CrossRef] [Green Version]
- Vásquez, C.; Lewis, D.L. The β2-adrenergic receptor specifically sequesters gs but signals through both Gs and Gi/o in rat sympathetic neurons. Neuroscience 2003, 118, 603–610. [Google Scholar] [CrossRef]
- Regard, J.B.; Cherman, N.; Palmer, D.; Kuznetsov, S.A.; Celi, F.S.; Guettier, J.M.; Chen, M.; Bhattacharyya, N.; Wess, J.; Coughlin, S.R.; et al. Wnt/beta-catenin signaling is differentially regulated by Galpha proteins and contributes to fibrous dysplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 20101–20106. [Google Scholar] [CrossRef] [Green Version]
- Reul, J.M.; de Kloet, E.R. Two Receptor Systems for Corticosterone in Rat Brain: Microdistribution and Differential Occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef] [PubMed]
- Sawtell, N.M.; Thompson, R.L. De Novo Herpes Simplex Virus VP16 Expression Gates a Dynamic Programmatic Transition and Sets the Latent/Lytic Balance during Acute Infection in Trigeminal Ganglia. PLoS Pathog. 2016, 12, e1005877. [Google Scholar] [CrossRef] [PubMed]
- Fryer, C.J.; Archer, T.K. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 1998, 393, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Marquart, M.E.; Zheng, X.; Tran, R.K.; Thompson, H.W.; Bloom, D.C.; Hill, J.M. A cAMP response element within the latency-associated transcript promoter of HSV-1 facilitates induced ocular reactivation in a mouse hyperthermia model. Virology 2001, 284, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Millhouse, S.; Kenny, J.J.; Quinn, P.G.; Lee, V.; Wigdahl, B. ATF/CREB elements in the herpes simplex virus type 1 latency-associated transcript promoter interact with members of the ATF/CREB and AP-1 transcription factor families. J. Biomed. Sci. 1998, 5, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hancock, M.; Workman, A.; Doster, A.; Jones, C. beta-Catenin, a Transcription Factor Activated by Canonical Wnt Signaling, Is Expressed in Sensory Neurons of Calves Latently Infected with Bovine Herpesvirus 1. J. Virol. 2016, 90, 3148–3159. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Thunuguntla, P.; Liu, Y.; Hancock, M.; Jones, C. The beta-catenin signaling pathway stimulates bovine herpesvirus 1 productive infection. Virology 2017, 500, 91–95. [Google Scholar] [CrossRef]
- Choi, E.J.; Kim, S.; Jho, E.H.; Song, K.J.; Kee, S.H. Axin expression enhances herpes simplex virus type 1 replication by inhibiting virus-mediated cell death in L929 cells. J. Gen. Virol. 2013, 94, 1636–1646. [Google Scholar] [CrossRef]
- Li, B.X.; Gardner, R.; Xue, C.; Qian, D.Z.; Xie, F.; Thomas, G.; Kazmierczak, S.C.; Habecker, B.A.; Xiao, X. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci. Rep. 2016, 6, 34513. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Lesiten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, J.; Zhang, J.; Pu, M.; Zhou, X.; Dong, L.; Mao, X.; Shi, G.; Zou, J.; Wu, J.; Jiang, D.; et al. Modulation of GSK-3beta/beta-Catenin Signaling Contributes to Learning and Memory Impairment in a Rat Model of Depression. Int. J. Neuropsychopharmacol. 2018, 21, 858–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S.; Nagourney, R.; Cardozo, T.; Brown, A.M.C.; DasGupta, R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, H.; Huang, S.; Qiu, Y. Histone deacetylase 1 and p300 can directly associate with chromatin and compete for binding in a mutually exclusive manner. PLoS ONE 2014, 9, e94523. [Google Scholar] [CrossRef]
- Sergerie, Y.; Boivin, G.; Gosselin, D.; Rivest, S. Delayed but not early glucocorticoid treatment protects the host during experimental herpes simplex virus encephalitis in mice. J. Infect. Dis. 2007, 195, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Hardwicke, M.A.; Schaffer, P.A. Differential effects of nerve growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. J. Virol. 1997, 71, 3580–3587. [Google Scholar] [CrossRef] [Green Version]
- Beyer, C.F.; Arens, M.Q.; Hill, J.M.; Rose, B.T.; Hill, G.A.; Lin, D.T. Penetrating keratoplasty in rabbits induces latent HSV-1 reactivation when corticosteroids are used. Curr. Eye Res. 1989, 8, 1323–1329. [Google Scholar] [CrossRef]
- Haruta, Y.; Rootman, D.S.; Xie, L.; Kiritoshi, A.; Hill, J.M. Recurrent HSV-1 corneal lesions in rabbits induced by cyclophosphamide and dexamethasone. Investig. Opthalmol. Vis. Sci. 1989, 30, 371–376. [Google Scholar]
- Jusufbegovic, D.; Schaal, S. Quiescent herpes simplex keratitis reactivation after intravitreal injection of dexamethasone implant. Retin. Cases Brief Rep. 2016, 11, 296–297. [Google Scholar] [CrossRef]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A CBP Integrator Complex Mediates Transcriptional Activation and AP-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Neumann, D.M.; Bhattacharjee, P.S.; Giordani, N.V.; Bloom, D.C.; Hill, J.M. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J. Virol. 2007, 81, 13248–13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, A.L.; Giordani, N.V.; Kubat, N.J.; O’Neil, J.E.; Bloom, D.C. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 2006, 80, 2063–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Kim, J.Y.; Camarena, V.; Roehm, P.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2013, 62, e3823. [Google Scholar] [CrossRef]
- Lee, S.; Ives, A.M.; Bertke, A.S. Herpes Simplex Virus 1 Reactivates from Autonomic Ciliary Ganglia Independently from Sensory Trigeminal Ganglia To Cause Recurrent Ocular Disease. J. Virol. 2015, 89, 8383–8391. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Kopin, I.J. Evolution of concepts of stress. Stress 2007, 10, 109–120. [Google Scholar] [CrossRef]
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goswami, P.; Ives, A.M.; Abbott, A.R.N.; Bertke, A.S. Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons. Viruses 2022, 14, 1115. https://doi.org/10.3390/v14051115
Goswami P, Ives AM, Abbott ARN, Bertke AS. Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons. Viruses. 2022; 14(5):1115. https://doi.org/10.3390/v14051115
Chicago/Turabian StyleGoswami, Poorna, Angela M. Ives, Amber R. N. Abbott, and Andrea S. Bertke. 2022. "Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons" Viruses 14, no. 5: 1115. https://doi.org/10.3390/v14051115
APA StyleGoswami, P., Ives, A. M., Abbott, A. R. N., & Bertke, A. S. (2022). Stress Hormones Epinephrine and Corticosterone Selectively Reactivate HSV-1 and HSV-2 in Sympathetic and Sensory Neurons. Viruses, 14(5), 1115. https://doi.org/10.3390/v14051115