
Detecting Potentially Adaptive Mutations from the Parallel and Fixed Patterns in SARS-CoV-2 Evolution

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
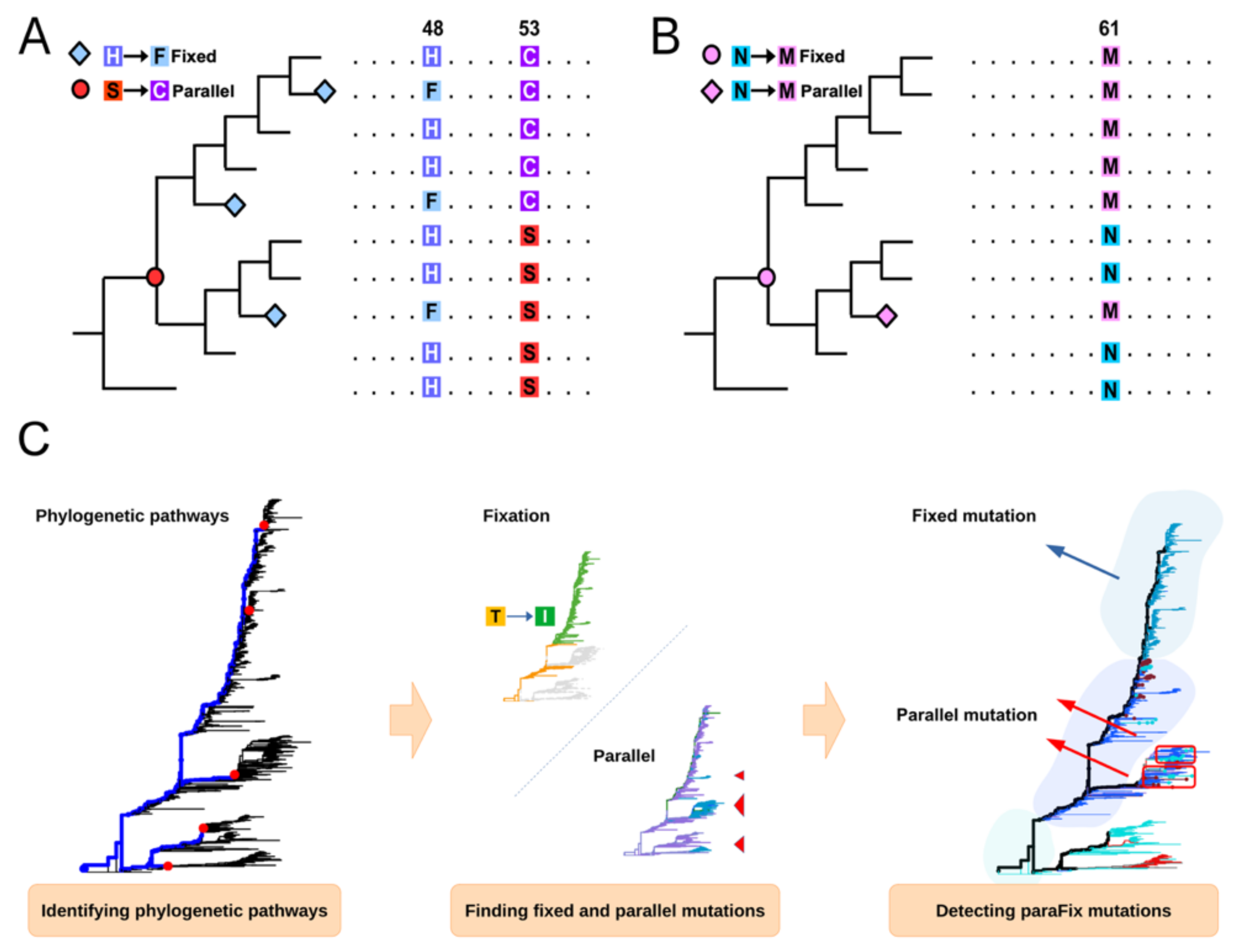
2.2. A Brief Review of SitePath
2.3. Detection of ParaFix, Homoplasy, and Episodic Positive Selection Sites
2.4. Comparison of Performance between SitePath and HyPhy MEME
3. Results
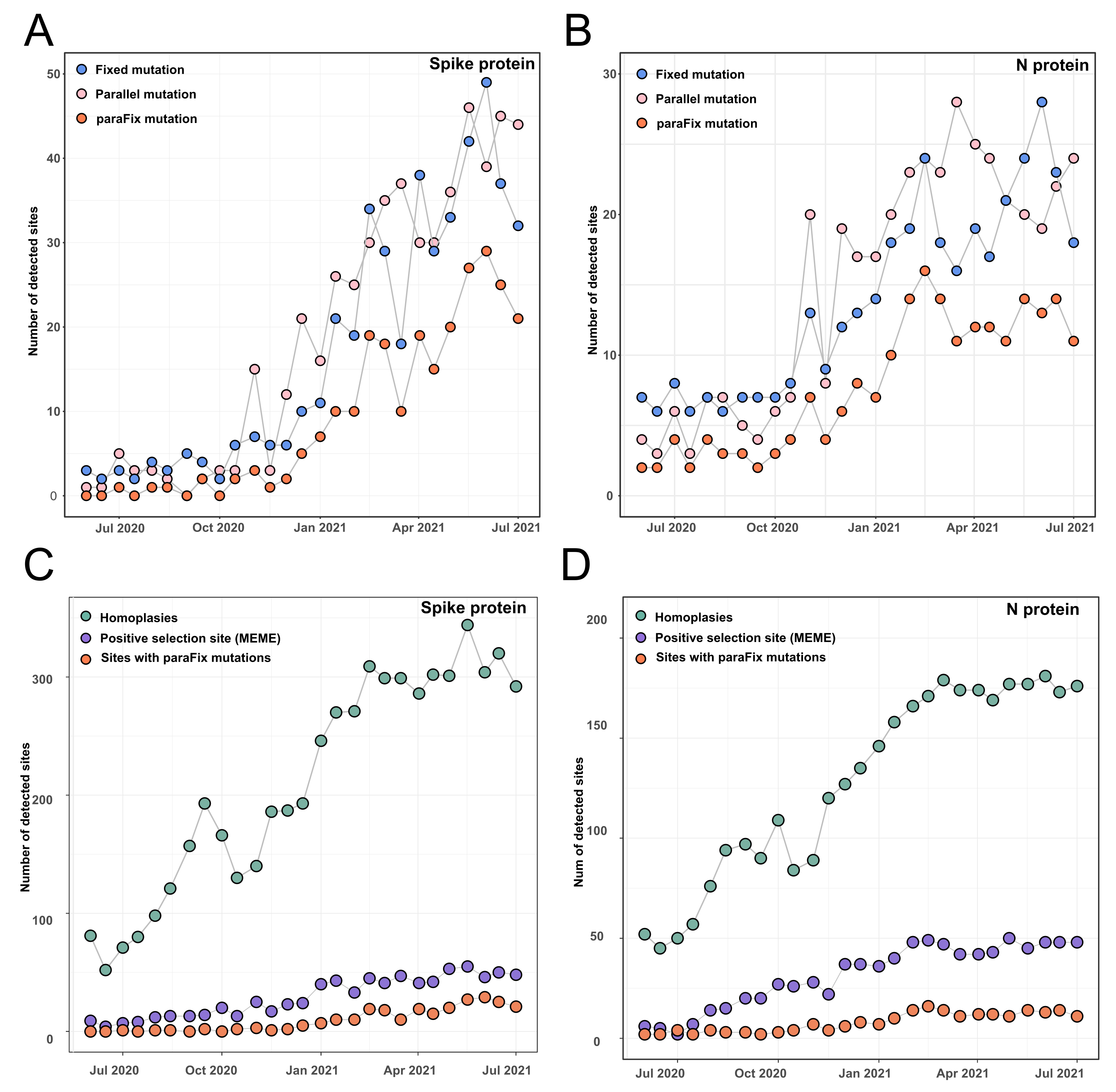
3.1. Tracking Dynamic Evolutionary Patterns of SARS-CoV-2
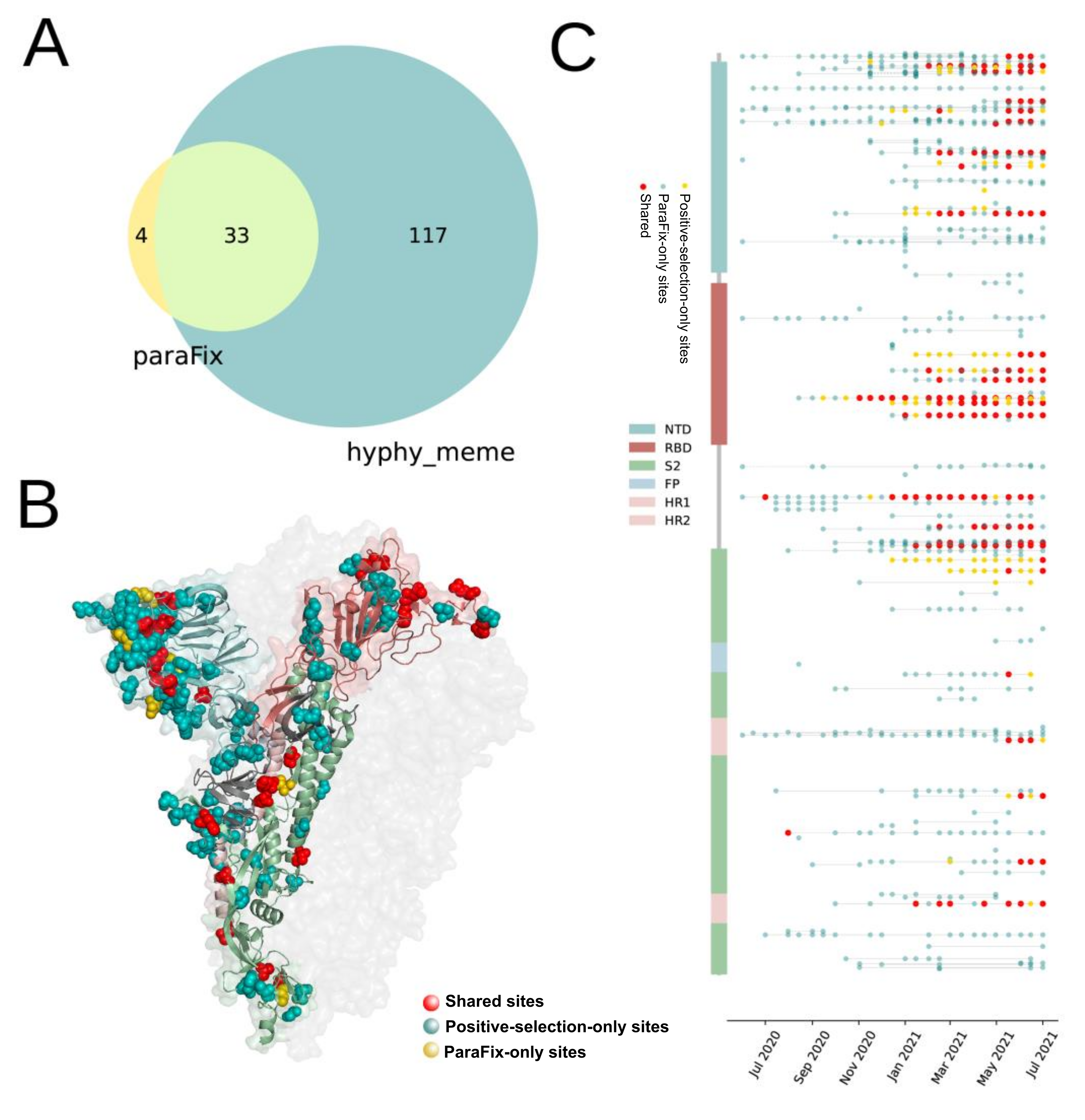
3.2. Comparison of Sites with ParaFix Mutation, Positive Selection Sites, and Homoplasies
3.3. Comparision of Sites with ParaFix Mutaiton and Positive Selective Sites in S Protein
3.4. ParaFix Sites as Indicators of Potentially Dominant SARS-CoV-2 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, L.; Richard, D.; Tan, C.C.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mapping the evolutionary potential of RNA viruses. Cell Host Microbe 2018, 23, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Bobay, L.-M.; Ochman, H. Impact of recombination on the base composition of bacteria and archaea. Mol. Biol. Evol. 2017, 34, 2627–2636. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Zhou, H.; Wu, A. sitePath: Phylogeny-Based Sequence Clustering with Site Polymorphism. R Package Version 1.10.2. Available online: https://wuaipinglab.github.io/sitePath/ (accessed on 30 April 2022).
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. In Statistical Methods in Molecular Evolution; Springer: Berlin/Heidelberg, Germany, 2005; pp. 125–181. [Google Scholar]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L. Increased resistance of SARS-CoV-2 variant P. 1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D. Antibody resistance of SARS-CoV-2 variants B. 1.351 and B. 1.1. 7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.E.; Dang, H.V.; De Marco, A.; Franko, N.; Tilles, S.W.; Logue, J. Molecular basis of immune evasion by the delta and kappa SARS-CoV-2 variants. Science 2021, 374, 1621–1626. [Google Scholar] [CrossRef]
- Cerutti, G.; Guo, Y.; Wang, P.; Nair, M.S.; Wang, M.; Huang, Y.; Yu, J.; Liu, L.; Katsamba, P.S.; Bahna, F. Neutralizing antibody 5-7 defines a distinct site of vulnerability in SARS-CoV-2 spike N-terminal domain. Cell Rep. 2021, 37, 109928. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Kuzmina, A.; Khalaila, Y.; Voloshin, O.; Keren-Naus, A.; Boehm-Cohen, L.; Raviv, Y.; Shemer-Avni, Y.; Rosenberg, E.; Taube, R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe 2021, 29, 522–528.e2. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, D.; Lee, C.-C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.; Tuekprakhon, A.; Nutalai, R. Evidence of escape of SARS-CoV-2 variant B. 1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348–2361.e46. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, M.E.; Haslwanter, D.; Bortz, I.I.I.R.H.; Wirchnianski, A.S.; Lasso, G.; Vergnolle, O.; Abbasi, S.A.; Fels, J.M.; Laudermilch, E.; Florez, C. A replication-competent vesicular stomatitis virus for studies of SARS-CoV-2 spike-mediated cell entry and its inhibition. Cell Host Microbe 2020, 28, 486–496.e6. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Evans, J.P.; Faraone, J.N.; Qu, P.; Zheng, Y.-M.; Saif, L.; Oltz, E.M.; Lozanski, G.; Gumina, R.J.; Liu, S.-L. Neutralization of SARS-CoV-2 variants of concern harboring Q677H. mBio 2021, 12, e02510-21. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Kosugi, Y.; Wu, J.; Zahradnik, J.; Yamasoba, D.; Butlertanaka, E.P.; Tanaka, Y.L.; Uriu, K.; Liu, Y.; Morizako, N. The SARS-CoV-2 Lambda variant exhibits enhanced infectivity and immune resistance. Cell Rep. 2022, 38, 110218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Functions | References |
|---|---|---|
| L5F | Enhanced infectivity with D614G, but decreased infectivity without D614G | [14] |
| S12F | Indirectly contributes to antibody escape | [14] |
| L18F | 1. Escape of antibody S2L28 2. L18F, T20N, and D138Y contributed to the loss of activity of samples 2–17 and samples 4–19 | [15,16] |
| T20N | L18F, T20N, and D138Y contributed to the loss of activity of samples 2–17 and samples 4–19 | [16] |
| P26S | Partially accounted for the loss of activity of samples 4–18 | [16] |
| A67V | - | - |
| D80A | 1. Slightly reduced the antibody neutralization of S2L28 2. High resistance to convalescent plasma of P6 and high sensitization to P18 | [16,17] |
| T95I | T95I substitution occurs outside the antigenic supersite and is unlikely to significantly contribute to immune evasion | [18] |
| S98F | - | - |
| D138Y | L18F, T20N, and D138Y contributed to the loss of activity of samples 2–17 and samples 4–19 | [16] |
| W152C | 1. Both R190S and W152C impair binding of samples 5–7 by altering the local conformation of NTD loops 2. R190S and W152C each attenuate the pseudovirus half-maximal inhibitory concentration (IC50) by 6.1- and 18.9-fold, respectively | [19] |
| E156G | Residues 156–158 participate in the supersite β-hairpin and their mutation/deletion in the B.1.617.2 NTD lead to striking structural remodeling | [18] |
| F157L | Residues 156–158 participate in the supersite β-hairpin and their mutation/deletion in the B.1.617.2 NTD lead to striking structural remodeling | [18] |
| R190S | 1. Both R190S and W152C impair binding of samples 5–7 by altering the local conformation of NTD loops 2. R190S and W152C each attenuate the pseudovirus half-maximal inhibitory concentration (IC50) by 6.1- and 18.9-fold, respectively 3. L18F, T20N, D138Y, and R190S together resulted in the loss of activity of samples 5–7 | [16,19] |
| D215G | High resistance to convalescent plasma of P6, P8, P11, P13, P15, P17 | [17] |
| A222V | Not strong for antibody escape | [15,20] |
| K417N/T | K417N seems to eliminate neutralization by co-occurrence with E484K | [21,22] |
| N439K | N439K increases the spike affinity for hACE2; viral fitness and disease are unchanged, N439K confers resistance to several mAbs and escapes some polyclonal responses | [23] |
| L452R/Q | The L452R mutation reduces neutralization mediated by some clinical mAbs, such as bamlanivimab (LY-CoV555) and regdanvimab (CT-P59), due to steric alteration of the antigenic site, which is incompatible with binding | [18,24] |
| S477N | No impact was observed on either mAb with the S477N or K537R variants | [25] |
| T478K | Requires further investigation | [18,26] |
| E484K/Q | Antibody escape | [27] |
| N501Y/T | Enhances SARS-CoV-2 infection and transmission | [28] |
| D614G | Increases viral infectivity | [29] |
| H655Y | Confers SARS-CoV-2 with competence priority | [30] |
| Q677H | Increases viral infectivity and syncytium formation, and enhances resistance to neutralization for VOCs | [31] |
| P681H/R | Increases viral infectivity | [32,33] |
| A701V | High resistance to convalescent plasma of P2 and P11 | [17] |
| T716I | High resistance to convalescent plasma of P6, P11, and P13 | [17] |
| T732A | - | - |
| T859N | No influence on infectivity or vaccine-induced neutralization | [34] |
| D950N | D950N substitutions are part of epitopes known to be recognized by neutralizing Abs | [18] |
| T1027I | No major changes in S2, and V1176F is in a disordered region | [35] |
| A1078S | - | - |
| T1117I | - | - |
| D1118H | 1. High resistance to convalescent plasma of P6, P8, P11, and P13. 2. Decreases viral fitness in hamster | [17,28] |
| V1176F | V1176F is in a disordered region. | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C.-Y.; Han, N.; Cheng, Y.-X.; Shang, J.; Weng, S.; Yang, R.; Zhou, H.-Y.; Wu, A. Detecting Potentially Adaptive Mutations from the Parallel and Fixed Patterns in SARS-CoV-2 Evolution. Viruses 2022, 14, 1087. https://doi.org/10.3390/v14051087
Ji C-Y, Han N, Cheng Y-X, Shang J, Weng S, Yang R, Zhou H-Y, Wu A. Detecting Potentially Adaptive Mutations from the Parallel and Fixed Patterns in SARS-CoV-2 Evolution. Viruses. 2022; 14(5):1087. https://doi.org/10.3390/v14051087
Chicago/Turabian StyleJi, Cheng-Yang, Na Han, Ye-Xiao Cheng, Jingzhe Shang, Shenghui Weng, Rong Yang, Hang-Yu Zhou, and Aiping Wu. 2022. "Detecting Potentially Adaptive Mutations from the Parallel and Fixed Patterns in SARS-CoV-2 Evolution" Viruses 14, no. 5: 1087. https://doi.org/10.3390/v14051087
APA StyleJi, C.-Y., Han, N., Cheng, Y.-X., Shang, J., Weng, S., Yang, R., Zhou, H.-Y., & Wu, A. (2022). Detecting Potentially Adaptive Mutations from the Parallel and Fixed Patterns in SARS-CoV-2 Evolution. Viruses, 14(5), 1087. https://doi.org/10.3390/v14051087