
Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Clinical Specimens during Sentinel Surveillance
2.2. Primer Design and cDNA Synthesis for Whole-Genome Amplification
2.3. Construction of Genomic Libraries and Library Sequencing
2.4. Bioinformatic Pipeline for CVA6 Genome Analysis
2.5. Phylogenetic Analyses of Complete Genomes and Open Reading Frames (ORF)
2.6. Recombination Analyses
2.7. Statistical Analysis
3. Results
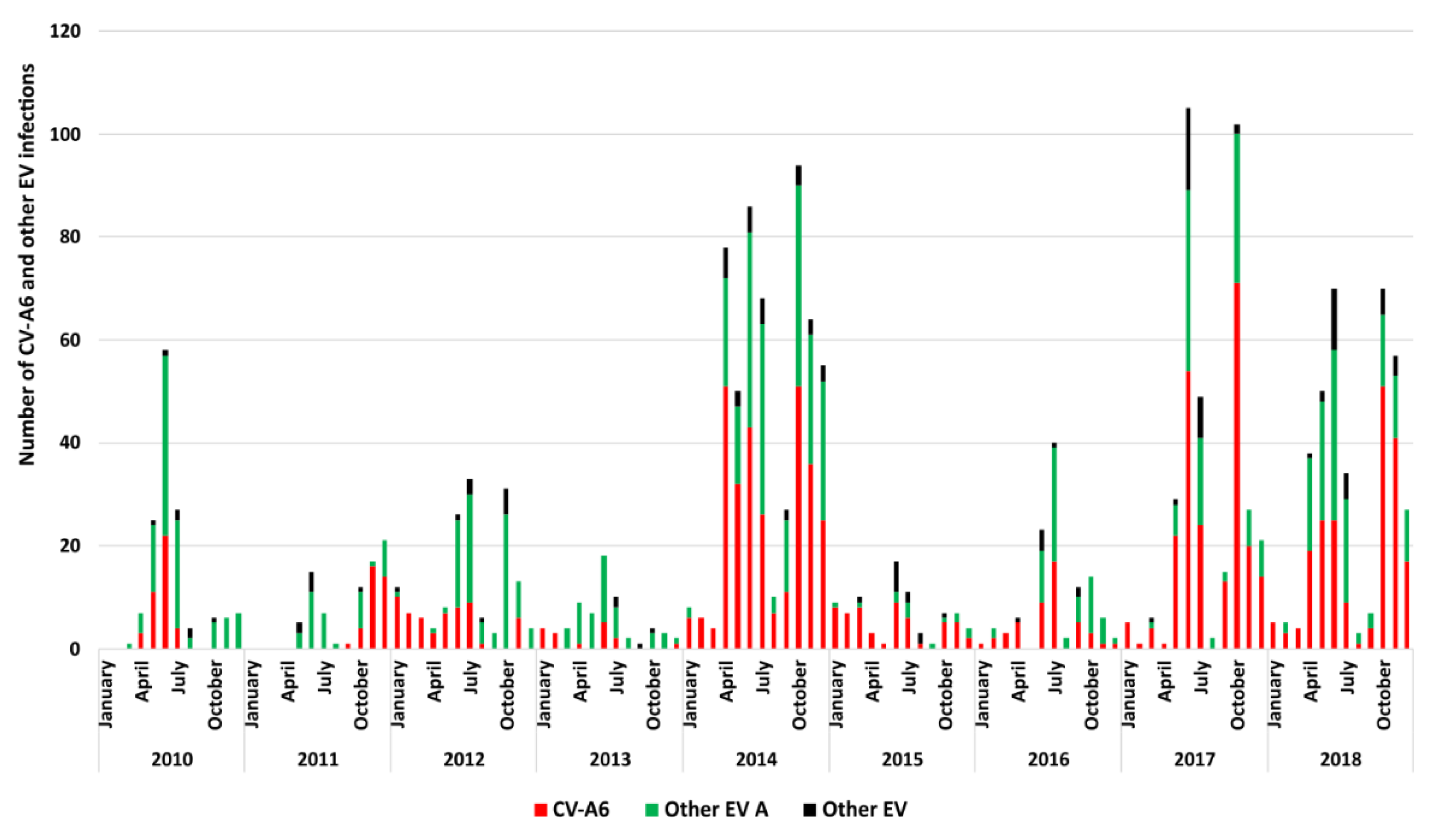
3.1. Epidemiologic and Clinical Characteristics of HFMD Cases Associated with CVA6 in France
3.2. Sequencing of CVA6 Complete Genomes from Clinical Samples
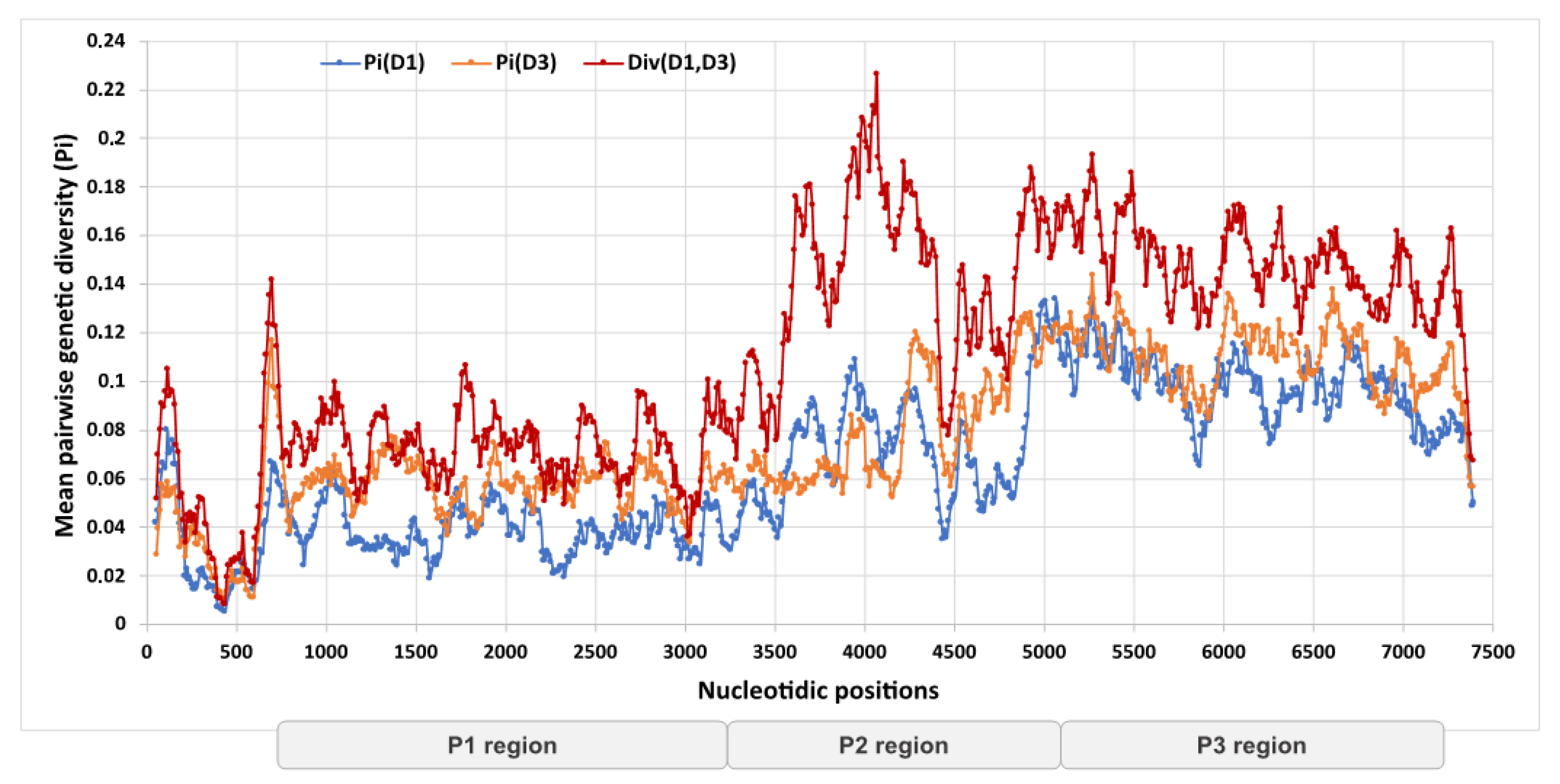
3.3. Genetic Divergence of Whole Genome Sequences and Assignment to Recombinant Forms
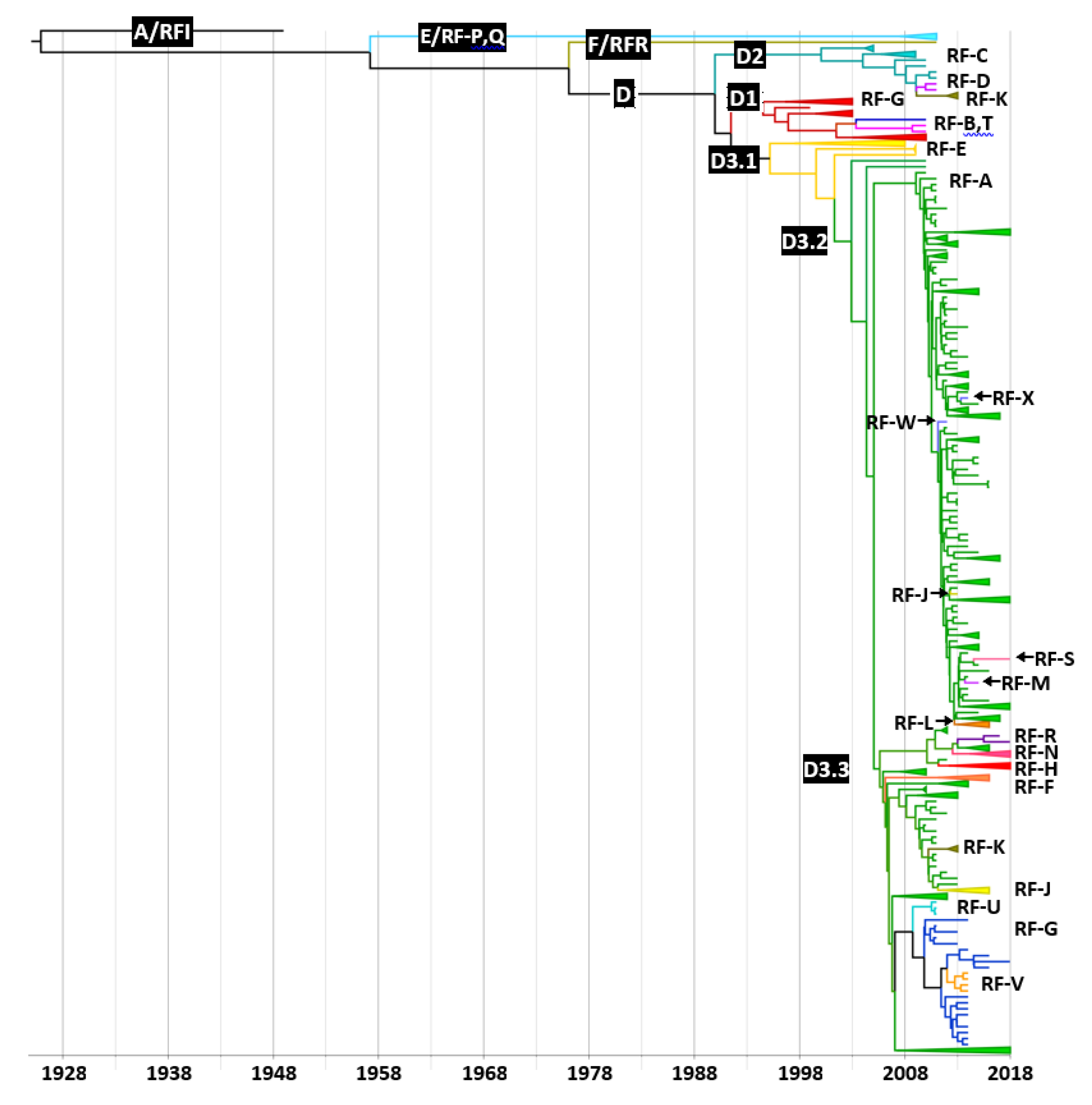
3.4. Distribution of Recombinant Forms among CVA6 Clades and Subclades
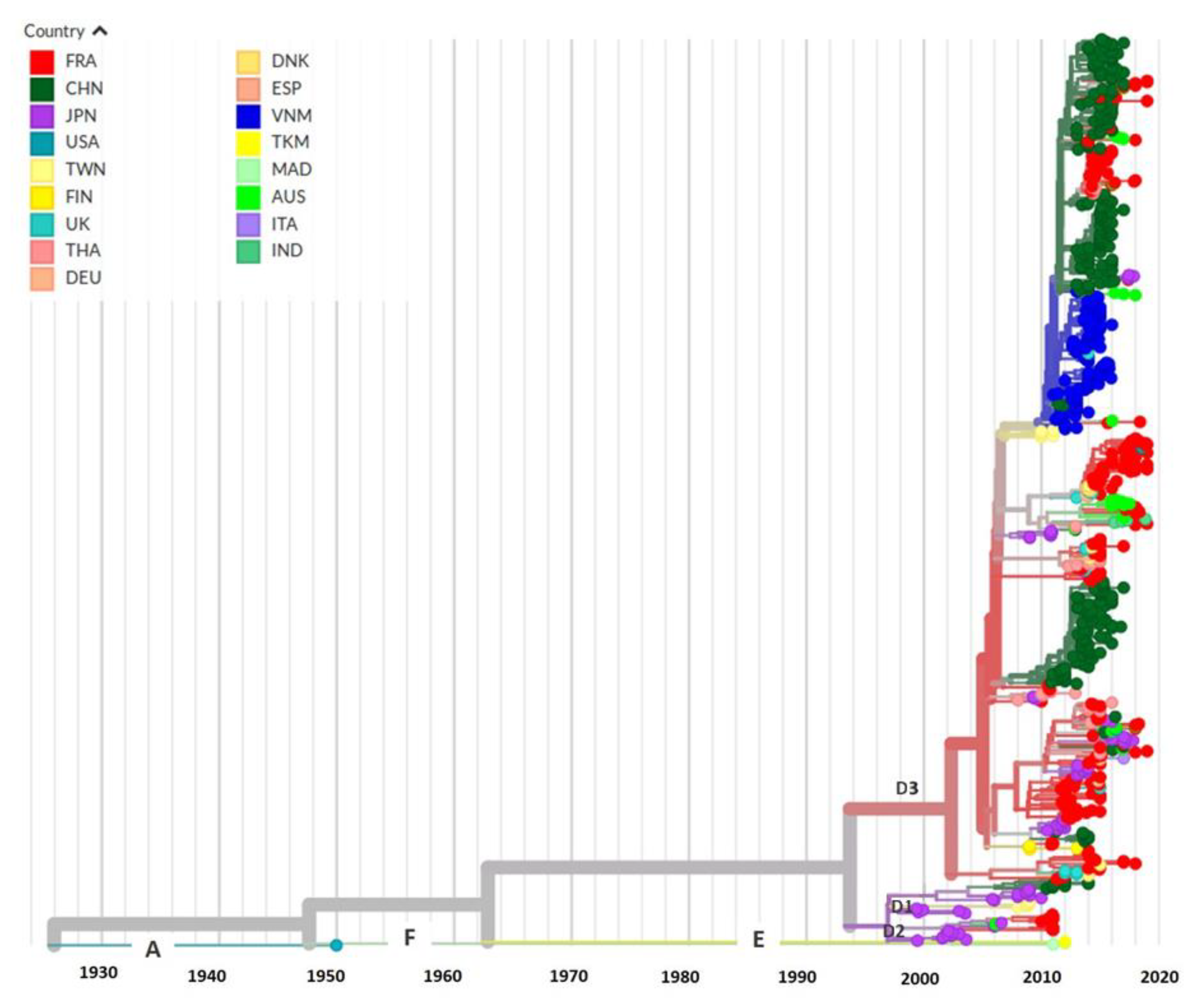
3.5. Evolutionary History and Geographic Distribution of CVA6
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osterback, R.; Vuorinen, T.; Linna, M.; Susi, P.; Hyypiä, T.; Waris, M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg. Infect. Dis. 2009, 15, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Cabrerizo, M.; Tarragó, D.; Muñoz-Almagro, C.; del Amo, E.; Domínguez-Gil, M.; Eiros, J.M.; López-Miragaya, I.; Pérez, C.; Reina, J.; Otero, A.; et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin. Microbiol. Infect. 2014, 20, O150–O156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Iizuka, S.; Enomoto, M.; Abe, K.; Yamashita, K.; Hanaoka, N.; Okabe, N.; Yoshida, H.; Yasui, Y.; Kobayashi, M.; et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg. Infect. Dis. 2012, 18, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Hayman, R.; Shepherd, M.; Tarring, C.; Best, E. Outbreak of variant hand-foot-and-mouth disease caused by coxsackievirus A6 in Auckland, New Zealand. J. Paediatr. Child Health 2014, 50, 751–755. [Google Scholar] [CrossRef]
- Cisterna, D.M.; Lema, C.L.; Martinez, L.M.; Verón, E.; Contarino, L.P.; Acosta, D.; Freire, M.C. Atypical hand, foot, and mouth disease caused by Coxsackievirus A6 in Argentina in 2015. Rev. Argent. Microbiol. 2019, 51, 140–143. [Google Scholar] [CrossRef]
- Bubba, L.; Broberg, E.K.; Jasir, A.; Simmonds, P.; Harvala, H.; Enterovirus study collaborators. Circulation of non-polio enteroviruses in 24 EU and EEA countries between 2015 and 2017: A retrospective surveillance study. Lancet Infect. Dis. 2020, 20, 350–361. [Google Scholar] [CrossRef]
- Martínez-López, N.; Muñoz-Almagro, C.; Launes, C.; Navascués, A.; Imaz-Pérez, M.; Reina, J.; Romero, M.P.; Calvo, C.; Ruiz-García, M.; Megias, G.; et al. Surveillance for Enteroviruses Associated with Hand, Foot, and Mouth Disease, and Other Mucocutaneous Symptoms in Spain, 2006–2020. Viruses 2021, 13, 781. [Google Scholar] [CrossRef]
- Mirand, A.; Cohen, R.; Bisseux, M.; Tomba, S.; Sellem, F.C.; Gelbert, N.; Béchet, S.; Frandji, B.; Archimbaud, C.; Brebion, A.; et al. A large-scale outbreak of hand, foot and mouth disease, France, as at 28 September 2021. Eurosurveillance 2021, 26, 2100978. [Google Scholar] [CrossRef]
- Mathes, E.F.; Oza, V.; Frieden, I.J.; Cordoro, K.M.; Yagi, S.; Howard, R.; Kristal, L.; Ginocchio, C.C.; Schaffer, J.; Maguiness, S.; et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics 2013, 132, e149–e157. [Google Scholar] [CrossRef] [Green Version]
- Feder, H.M., Jr.; Bennett, N.; Modlin, J.F. Atypical hand, foot, and mouth disease: A vesiculobullous eruption caused by Coxsackie virus A6. Lancet Infect. Dis. 2014, 14, 83–86. [Google Scholar] [CrossRef]
- Sinclair, C.; Gaunt, E.; Simmonds, P.; Broomfield, D.; Nwafor, N.; Wellington, L.; Templeton, K.; Willocks, L.; Schofield, O.; Harvala, H. Atypical hand, foot, and mouth disease associated with coxsackievirus A6 infection, Edinburgh, United Kingdom, January to February 2014. Eurosurveillance 2014, 19, 20745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, S.; Klemola, P.; Kaijalainen, S.; Paananen, A.; Simonen, M.L.; Vuorinen, T.; Roivainen, M. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J. Clin. Virol. 2010, 48, 49–54. [Google Scholar] [CrossRef]
- Balestri, R.; Bellino, M.; Landini, L.; Tasin, L.; Rizzoli, L.; Speziali, L.; Bauer, P.; Sicher, M.C.; Rech, G.; Girardelli, C.R. Atypical presentation of enterovirus infection in adults: Outbreak of ‘hand, foot, mouth and scalp disease’ in Northern Italy. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e60–e61. [Google Scholar] [CrossRef] [PubMed]
- Broccolo, F.; Drago, F.; Ciccarese, G.; Genoni, A.; Puggioni, A.; Rosa, G.M.; Parodi, A.; Manukyan, H.; Laassri, M.; Chumakov, K.; et al. Severe atypical hand-foot-and-mouth disease in adults due to coxsackievirus A6: Clinical presentation and phylogenesis of CV-A6 strains. J. Clin. Virol. 2019, 110, 1–6. [Google Scholar] [CrossRef]
- Yang, F.; Yuan, J.; Wang, X.; Li, J.; Du, J.; Su, H.; Zhou, B.; Jin, Q. Severe hand, foot, and mouth disease and coxsackievirus A6-Shenzhen, China. Clin. Infect. Dis. 2014, 59, 1504–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, L.; Wang, Y.; Yao, X.; Mao, Q.; Xu, M.; Liang, Z. Coxsackievirus A6: A new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev. Anti-Infect. Ther. 2015, 13, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X.; et al. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci. Rep. 2017, 7, 5491. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Zhao, P.S.H.; Sridhar, S.; Yip, C.C.Y.; Aw-Yong, K.L.; Chow, E.Y.Y.; Cheung, K.C.M.; Hui, R.W.H.; Leung, R.Y.H.; Lai, Y.S.K.; et al. Molecular epidemiology of coxsackievirus A6 circulating in Hong Kong reveals common neurological manifestations and emergence of novel recombinant groups. J. Clin. Virol. 2018, 108, 43–49. [Google Scholar] [CrossRef]
- Anh, N.T.; Nhu, L.N.T.; Van, H.M.T.; Hong, N.T.T.; Thanh, T.T.; Hang, V.T.T.; Ny, N.T.H.; Nguyet, L.A.; Phuong, T.T.L.; Nhan, L.N.T.; et al. Emerging Coxsackievirus A6 Causing Hand, Foot and Mouth Disease, Vietnam. Emerg. Infect. Dis. 2018, 24, 654–662. [Google Scholar] [CrossRef] [Green Version]
- Andrés, C.; Guasch, E.; Piñana, M.; Fernandes, P.; Gimferrer, L.; Esso, D.V.; Codina, M.G.; Esperalba, J.; Vila, J.; Rodrigo, C.; et al. Recombinant CV-A6 strains related to hand-foot-mouth disease and herpangina at primary care centers (Barcelona, Spain). Future Microbiol. 2019, 14, 499–507. [Google Scholar] [CrossRef]
- Gaunt, E.; Harvala, H.; Österback, R.; Sreenu, V.B.; Thomson, E.; Waris, M.; Simmonds, P. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: A potential role of recombination in emergence and pathogenicity. J. Gen. Virol. 2015, 96, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puenpa, J.; Vongpunsawad, S.; Österback, R.; Waris, M.; Eriksson, E.; Albert, J.; Midgley, S.; Fischer, T.K.; Eis-Hübinger, A.M.; Cabrerizo, M.; et al. Molecular epidemiology and the evolution of human coxsackievirus A6. J. Gen. Virol. 2016, 97, 3225–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhang, Y.; Han, Z.; Xu, W.; Xiao, J.; Wang, X.; Wang, J.; Yang, J.; Yu, Q.; Yu, D.; et al. Genetic recombination in fast-spreading coxsackievirus A6 variants: A potential role in evolution and pathogenicity. Virus Evol. 2020, 6, veaa048. [Google Scholar] [CrossRef] [PubMed]
- Mirand, A.; Henquell, C.; Archimbaud, C.; Ughetto, S.; Antona, D.; Bailly, J.L.; Peigue-Lafeuille, H. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 and A10 infections in 2010, France: A large citywide, prospective observational study. Clin. Microbiol. Infect. 2012, 18, E110–E118. [Google Scholar] [CrossRef] [Green Version]
- Mirand, A.; le Sage, F.V.; Pereira, B.; Cohen, R.; Levy, C.; Archimbaud, C.; Peigue-Lafeuille, H.; Bailly, J.L.; Henquell, C. Ambulatory Pediatric Surveillance of Hand, Foot and Mouth Disease as Signal of an Outbreak of Coxsackievirus A6 Infections, France, 2014–2015. Emerg. Infect. Dis. 2016, 22, 1884–1893. [Google Scholar] [CrossRef]
- Mirand, A.; Henquell, C.; Archimbaud, C.; Chambon, M.; Charbonné, F.; Peigue-Lafeuille, H.; Bailly, J.L. Prospective identification of enteroviruses involved in meningitis in 2006 through direct genotyping in cerebrospinal fluid. J. Clin. Microbiol. 2008, 46, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Ngangas, S.T.; Lukashev, A.; Jugie, G.; Ivanova, O.; Mansuy, J.M.; Mengelle, C.; Izopet, J.; L’honneur, A.S.; Rozenberg, F.; Leyssene, D.; et al. Multirecombinant Enterovirus A71 Subgenogroup C1 Isolates Associated with Neurologic Disease, France, 2016–2017. Emerg. Infect. Dis. 2019, 25, 1204–1208. [Google Scholar] [CrossRef]
- Cobbin, J.C.A.; Britton, P.N.; Burrell, R.; Thosar, D.; Selvakumar, K.; Eden, J.S.; Jones, C.A.; Holmes, E.C. A complex mosaic of enteroviruses shapes community-acquired hand, foot and mouth disease transmission and evolution within a single hospital. Virus Evol. 2018, 4, vey020. [Google Scholar] [CrossRef]
- Mizuta, K.; Tanaka, S.; Komabayashi, K.; Aoki, Y.; Itagaki, T.; Katsushima, F.; Katsushima, Y.; Yoshida, H.; Ito, S.; Matsuzaki, Y.; et al. Phylogenetic and antigenic analyses of coxsackievirus A6 isolates in Yamagata, Japan between 2001 and 2017. Vaccine 2019, 37, 1109–1117. [Google Scholar] [CrossRef]
- Isaacs, S.R.; Kim, K.W.; Cheng, J.X.; Bull, R.A.; Stelzer-Braid, S.; Luciani, F.; Rawlinson, W.D.; Craig, M.E. Amplification and next generation sequencing of near full-length human enteroviruses for identification and characterisation from clinical samples. Sci. Rep. 2018, 8, 11889. [Google Scholar] [CrossRef] [Green Version]
- Bessaud, M.; Razafindratsimandresy, R.; Nougairède, A.; Joffret, M.L.; Deshpande, J.M.; Dubot-Pérès, A.; Héraud, J.M.; de Lamballerie, X.; Delpeyroux, F.; Bailly, J.L. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS ONE 2014, 9, e90624. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Shumilina, E.Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Rouzine, I.M.; Bianco, S.; Acevedo, A.; Goldstein, E.F.; Farkov, M.; Brodsky, L.; Andino, R. RNA Recombination Enhances Adaptability and Is Required for Virus Spread and Virulence. Cell Host Microbe 2016, 19, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, S.; Obermeier, P.E.; Neubauer, K.; Diedrich, S.; Laboratory Network for Enterovirus Diagnostics. Recombinant Enterovirus A71 Subgenogroup C1 Strains, Germany, 2015. Emerg. Infect. Dis. 2016, 22, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.P.; Lin, X.D.; Chen, Y.P.; Liu, Q.; Wang, W.; Wang, C.Q.; Li, M.H.; Sun, X.Y.; Shi, M.; Holmes, E.C.; et al. Fourteen types of co-circulating recombinant enterovirus were associated with hand, foot, and mouth disease in children from Wenzhou, China. J. Clin. Virol. 2015, 70, 29–38. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Pons-Salort, M.; Grassly, N.C. Serotype-specific immunity explains the incidence of diseases caused by human enteroviruses. Science 2018, 361, 800–803. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Features a | All CVA6 Infections (n = 981) | 2010 (n = 40) | 2011 (n = 35) | 2012 (n = 57) | 2013 (n = 16) | 2014 (n = 298) | 2015 (n = 55) | 2016 (n = 47) | 2017 (n = 229) | 2018 (n = 204) | p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (year) b | 1.52 (1.05–2.21) | 1.76 (1.17–2.72) | 1.80 (1.25–2.38) | 1.58 (1–2.24) | 2.27 (1.33–3.48) | 1.56 (1.05–2.19) | 1.47 (1–1.95) | 1.51 (1.17–2.34) | 1.48 (1.03–2.03) | 1.46 (1–2.2) | 0.14 |
| Female | 408 (42) | 10 (25) | 19 (54.3) | 21 (36.8) | 8 (50) | 139 (47) | 18 (34) | 15 (32.6) | 83 (36.7) | 95 (46.8) | 0.02 |
| Male | 564 (58) | 30 (75) | 16 (45.7) | 36 (63.2) | 8 (50) | 157 (53) | 35 (66) | 31 (67.4) | 143 (63.3) | 108 (53.2) | |
| Herpangina | 57 (6) | 5 (12.5) | 4 (11.8) | 5 (8.8) | 1 (6.2) | 24 (8.1) | 1 (1.8) | 2 (4.3) | 11 (4.8) | 4 (2.0) | <0.001 |
| Typical HFMD | 214 (22) | 16 (40) | 8 (23.5) | 23 (40.3) | 3 (18.8) | 79 (26.7) | 18 (32.7) | 3 (6.4) | 31 (13.7) | 33 (16.2) | |
| Atypical HFMD | 705 (72) | 19 (47.5) | 22 (64.7) | 29 (50.9) | 12 (75) | 193 (65.2) | 36 (65.5) | 42 (89.3) | 185 (81.5) | 167 (81.8) |
| Sequencing Data | Library 1 | Library 2 | Library 3 |
|---|---|---|---|
| Generation of circular consensus sequences (CCS) | |||
| Number of CCS reads | 505,649 | 521,730 | 665,105 |
| Number of « subreads » | 4,571,660 | 3,608,692 | 5,319,387 |
| Median number of « subreads » per read | 9 | 6.9 | 8 |
| Results after filtering the subreads | |||
| Number of CCS | 201,359 | 162,812 | 274,118 |
| Number of « subreads » | 3,787,027 | 2,790,358 | 4,104,179 |
| Mean number of « subreads » per CCS | 18.8 | 17.1 | 15 |
| Results after demultiplexing | |||
| Number of CCS assigned to the barcodes attributed to samples | 108,510 | 88,608 | 171,991 |
| Mean number of CCS per barcode | 1790 | 1748 | 1864 |
| Number of viral genomes obtained | 95 | 95 | 96 |
| Mean number of nucleotide sequences per sample | 1066 | 865 | 1709 |
| Analysed samples | |||
| Coxsackievirus A6 | 81 | 96 | 49 |
| Enterovirus A71 a | 15 | - | - |
| Other types or control a | - | - | 47 |
| RF | Number of Viral Genomes (n = 213) | 2010 (n = 19) | 2011 (n = 9) | 2012 (n = 15) | 2013 (n = 4) | 2014 (n = 86) | 2015 (n = 21) | 2016 (n = 12) | 2017 (n = 18) | 2018 (n = 29) | Recombinant Lineage (Clade/RF) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 123 (58) | 9 (47.4) | 6 (66.7) | 15 (100) | 3 (75) | 58 (67.4) | 14 (67) | 3 (25) | 8 (44.4) | 7 (24.1) | D3/A |
| B | 7 (3.3) | 7 (36.8) | - | - | - | - | - | - | - | - | D1/B |
| C | 0 | - | - | - | - | - | - | - | - | - | D2/C |
| D | 0 | - | - | - | - | - | - | - | - | - | D2/D |
| E | 0 | - | - | - | - | - | - | - | - | - | D3/E |
| F | 11 (5.2) | - | - | - | - | 9 (10.5) | 1 (4) | 1 (8.3) | - | - | D3/F |
| G | 13 (6.1) | 1 (5.3) | - | - | 1 (25) | 8 (9.3) | - | 2 (16.7) | - | 1 (3.4) | D1G; D3/G |
| H | 41 (19) | - | - | - | - | 7 (8.1) | 6 (29) | 6 (50) | 8 (44.4) | 14 (48.3) | D3/H |
| I | 0 | - | - | - | - | - | - | - | - | - | A/I |
| J | 0 | - | - | - | - | - | - | - | - | - | D3/J |
| K | 0 | - | - | - | - | - | - | - | - | - | D2K; D3/K |
| L | 0 | - | - | - | - | - | - | - | - | - | D3/L |
| M | 0 | - | - | - | - | - | - | - | - | - | D3/M |
| N | 6 (2.8) | - | - | - | - | - | - | - | 1 (5.6) | 5 (17.4) | D3/N |
| P | 0 | - | - | - | - | - | - | - | - | - | E/P |
| Q | 0 | - | - | - | - | - | - | - | - | - | E/Q |
| R | 2 (0.9) | - | - | - | - | - | - | - | 1 (5.6) | 1 (3.4) | D3/R; F/R |
| S | 1 (0.5) | - | - | - | - | - | - | - | - | 1 (3.4) | D3/S |
| T | 2 (0.9) | 2 (10.5) | - | - | - | - | - | - | - | - | D1/T |
| U | 3 (1.4) | - | 3 (33.3) | - | - | - | - | - | - | - | D3/U |
| V | 4 (1.9) | - | - | - | - | 4 (4.7) | - | - | - | - | D3/V |
| W | 0 | - | - | - | - | - | - | - | - | - | D3/W |
| X | 0 | - | - | - | - | - | - | - | - | - | D3/X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomba Ngangas, S.; Bisseux, M.; Jugie, G.; Lambert, C.; Cohen, R.; Werner, A.; Archimbaud, C.; Henquell, C.; Mirand, A.; Bailly, J.-L. Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018. Viruses 2022, 14, 1078. https://doi.org/10.3390/v14051078
Tomba Ngangas S, Bisseux M, Jugie G, Lambert C, Cohen R, Werner A, Archimbaud C, Henquell C, Mirand A, Bailly J-L. Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018. Viruses. 2022; 14(5):1078. https://doi.org/10.3390/v14051078
Chicago/Turabian StyleTomba Ngangas, Stéphanie, Maxime Bisseux, Gwendoline Jugie, Céline Lambert, Robert Cohen, Andreas Werner, Christine Archimbaud, Cécile Henquell, Audrey Mirand, and Jean-Luc Bailly. 2022. "Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018" Viruses 14, no. 5: 1078. https://doi.org/10.3390/v14051078
APA StyleTomba Ngangas, S., Bisseux, M., Jugie, G., Lambert, C., Cohen, R., Werner, A., Archimbaud, C., Henquell, C., Mirand, A., & Bailly, J.-L. (2022). Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018. Viruses, 14(5), 1078. https://doi.org/10.3390/v14051078