
Tumors and Cytomegalovirus: An Intimate Interplay

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
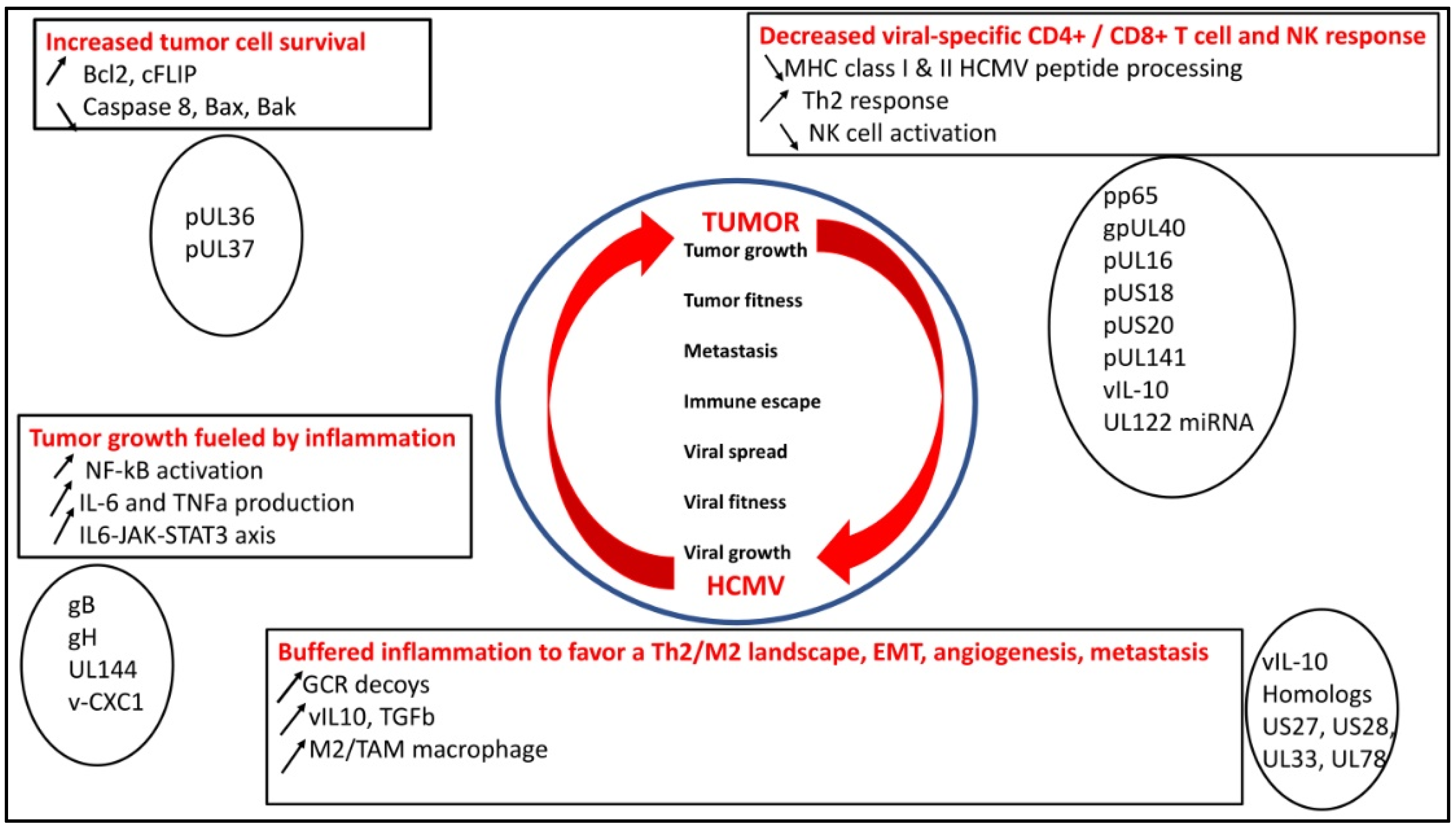
2. HCMV and Tumor, a “Win–Win” Strategy
2.1. HCMV Decreases Viral-Specific CD4+ and CD8+ T-Cell Response, NK Activity, which Could Favor Tumor Growth
2.2. HCMV Favors Tumor Cell Survival
2.3. HCMV Buffers the Inflammatory Landscape and Favors a TH2/M2 Landscape
3. HCMV Oncomodulation
4. HCMV Oncogenesis and the PGCC Paradigm
5. Antiviral Therapy and Immunotherapy Used to Fight HCMV-Linked Tumors
6. Conclusions
Funding
Conflicts of Interest
References
- Solana, R.; Tarazona, R.; Aiello, A.E.; Akbar, A.N.; Appay, V.; Beswick, M.; Bosch, J.A.; Campos, C.; Cantisán, S.; Cicin-Sain, L.; et al. CMV and Immunosenescence: From Basics to Clinics. Immun. Ageing 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- El Baba, R.; Herbein, G. Immune Landscape of CMV Infection in Cancer Patients: From “Canonical” Diseases toward Virus-Elicited Oncomodulation. Front. Immunol. 2021, 12, 730765. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.; Kartikasari, A.E.R.; Gorry, P.R.; Flanagan, K.L.; Plebanski, M. Potential Impact of Human Cytomegalovirus Infection on Immunity to Ovarian Tumours and Cancer Progression. Biomedicines 2021, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Scholz, M.; Doerr, H.W. Role of Tumor Cell Immune Escape Mechanisms in Cytomegalovirus-Mediated Oncomodulation. Med. Res. Rev. 2005, 25, 167–185. [Google Scholar] [CrossRef]
- Söderberg-Nauclér, C.; Geisler, J.; Vetvik, K. The Emerging Role of Human Cytomegalovirus Infection in Human Carcinogenesis: A Review of Current Evidence and Potential Therapeutic Implications. Oncotarget 2019, 10, 4333–4347. [Google Scholar] [CrossRef]
- Herbein, G. The Human Cytomegalovirus, from Oncomodulation to Oncogenesis. Viruses 2018, 10, 408. [Google Scholar] [CrossRef]
- Pasquereau, S.; Al Moussawi, F.; Karam, W.; Diab Assaf, M.; Kumar, A.; Herbein, G. Cytomegalovirus, Macrophages and Breast Cancer. Open Virol. J. 2017, 11, 15–27. [Google Scholar] [CrossRef]
- Touma, J.; Liu, Y.; Rahbar, A.; Pantalone, M.R.; Almazan, N.M.; Vetvik, K.; Söderberg-Nauclér, C.; Geisler, J.; Sauer, T. Detection of Human Cytomegalovirus Proteins in Paraffin-Embedded Breast Cancer Tissue Specimens-A Novel, Automated Immunohistochemical Staining Protocol. Microorganisms 2021, 9, 1059. [Google Scholar] [CrossRef]
- Peredo-Harvey, I.; Rahbar, A.; Söderberg-Nauclér, C. Presence of the Human Cytomegalovirus in Glioblastomas-A Systematic Review. Cancers 2021, 13, 5051. [Google Scholar] [CrossRef]
- Herbein, G.; Nehme, Z. Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge. Front. Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef]
- Shen, Y.; Zhu, H.; Shenk, T. Human Cytomegalovirus IE1 and IE2 Proteins Are Mutagenic and Mediate “Hit-and-Run” Oncogenic Transformation in Cooperation with the Adenovirus E1A Proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 3341–3345. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Cobbs, C.S. Is HCMV a Tumor Promoter? Virus Res. 2011, 157, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.; Touma, J.; Rahbar, A.; Söderberg-Nauclér, C.; Vetvik, K. A Review of the Potential Role of Human Cytomegalovirus (HCMV) Infections in Breast Cancer Carcinogenesis and Abnormal Immunity. Cancers 2019, 11, 1842. [Google Scholar] [CrossRef] [PubMed]
- Lepiller, Q.; Aziz Khan, K.; Di Martino, V.; Herbein, G. Cytomegalovirus and Tumors: Two Players for One Goal-Immune Escape. Open Virol. J. 2011, 5, 60–69. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Haidar Ahmad, S.; Coaquette, A.; Molimard, C.; Monnien, F.; Algros, M.-P.; Adotevi, O.; Diab Assaf, M.; Feugeas, J.-P.; et al. Polyploid Giant Cancer Cells, Stemness and Epithelial-Mesenchymal Plasticity Elicited by Human Cytomegalovirus. Oncogene 2021, 40, 3030–3046. [Google Scholar] [CrossRef]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid Giant Cancer Cells: An Emerging New Field of Cancer Biology. Semin. Cancer Biol. 2021, 81, 1–4. [Google Scholar] [CrossRef]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer Recurrence and Lethality Are Enabled by Enhanced Survival and Reversible Cell Cycle Arrest of Polyaneuploid Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118. [Google Scholar] [CrossRef]
- Liu, J.; Niu, N.; Li, X.; Zhang, X.; Sood, A.K. The Life Cycle of Polyploid Giant Cancer Cells and Dormancy in Cancer: Opportunities for Novel Therapeutic Interventions. Semin. Cancer Biol. 2022, 81, 132–144. [Google Scholar] [CrossRef]
- Müller-Coan, B.G.; Caetano, B.F.R.; Pagano, J.S.; Elgui de Oliveira, D. Cancer Progression Goes Viral: The Role of Oncoviruses in Aggressiveness of Malignancies. Trends Cancer 2018, 4, 485–498. [Google Scholar] [CrossRef]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human Cytomegalovirus Activates Inflammatory Cytokine Responses via CD14 and Toll-Like Receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef]
- Jackson, S.E.; Mason, G.M.; Wills, M.R. Human Cytomegalovirus Immunity and Immune Evasion. Virus Res. 2011, 157, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Herbein, G. Epigenetic Regulation of Human Cytomegalovirus Latency: An Update. Epigenomics 2014, 6, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive Genome-Wide Variability of Human Cytomegalovirus in Congenitally Infected Infants. PLoS Pathog 2011, 7, e1001344. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, A.; Götting, J.; Varanasi, P.R.; Steinbrueck, L.; Camiolo, S.; Zischke, J.; Heim, A.; Schulz, T.F.; Weissinger, E.M.; Kay-Fedorov, P.C.; et al. Human Cytomegalovirus Multiple-Strain Infections and Viral Population Diversity in Haematopoietic Stem Cell Transplant Recipients Analysed by High-Throughput Sequencing. Med. Microbiol. Immunol. 2021, 210, 291–304. [Google Scholar] [CrossRef]
- Götting, J.; Lazar, K.; Suárez, N.M.; Steinbrück, L.; Rabe, T.; Goelz, R.; Schulz, T.F.; Davison, A.J.; Hamprecht, K.; Ganzenmueller, T. Human Cytomegalovirus Genome Diversity in Longitudinally Collected Breast Milk Samples. Front. Cell Infect. Microbiol. 2021, 11, 664247. [Google Scholar] [CrossRef]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human Cytomegalovirus Genomes Sequenced Directly from Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef]
- Huang, E.S.; Alford, C.A.; Reynolds, D.W.; Stagno, S.; Pass, R.F. Molecular Epidemiology of Cytomegalovirus Infections in Women and Their Infants. N. Engl. J. Med. 1980, 303, 958–962. [Google Scholar] [CrossRef]
- Puchhammer-Stöckl, E.; Görzer, I. Cytomegalovirus and Epstein-Barr Virus Subtypes--the Search for Clinical Significance. J. Clin. Virol. 2006, 36, 239–248. [Google Scholar] [CrossRef]
- Coaquette, A.; Bourgeois, A.; Dirand, C.; Varin, A.; Chen, W.; Herbein, G. Mixed Cytomegalovirus Glycoprotein B Genotypes in Immunocompromised Patients. Clin. Infect. Dis. 2004, 39, 155–161. [Google Scholar] [CrossRef]
- Sowmya, P.; Madhavan, H.N. Analysis of Mixed Infections by Multiple Genotypes of Human Cytomegalovirus in Immunocompromised Patients. J. Med. Virol. 2009, 81, 861–869. [Google Scholar] [CrossRef]
- Hasing, M.E.; Pang, X.L.; Mabilangan, C.; Preiksaitis, J.K. Donor Cytomegalovirus Transmission Patterns in Solid Organ Transplant Recipients with Primary Infection. J. Infect. Dis. 2021, 223, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.M.; Blyth, E.; Li, K.; Ganzenmueller, T.; Camiolo, S.; Avdic, S.; Withers, B.; Linnenweber-Held, S.; Gwinner, W.; Dhingra, A.; et al. Whole-Genome Approach to Assessing Human Cytomegalovirus Dynamics in Transplant Patients Undergoing Antiviral Therapy. Front. Cell Infect. Microbiol. 2020, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Görzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stöckl, E. Deep Sequencing Reveals Highly Complex Dynamics of Human Cytomegalovirus Genotypes in Transplant Patients over Time. J. Virol. 2010, 84, 7195–7203. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PLoS Genet. 2013, 9, e1003735. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly from Clinical Specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef]
- Sijmons, S.; Thys, K.; Mbong Ngwese, M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.G.; et al. High-Resolution Human Cytomegalovirus Transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef]
- Wilkinson, G.W.G.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.Y.; Weekes, M.; Lehner, P.J.; et al. Human Cytomegalovirus: Taking the Strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar] [CrossRef]
- Xu, S.; Schafer, X.; Munger, J. Expression of Oncogenic Alleles Induces Multiple Blocks to Human Cytomegalovirus Infection. J. Virol. 2016, 90, 4346–4356. [Google Scholar] [CrossRef]
- Haidar Ahmad, S.; Pasquereau, S.; El Baba, R.; Nehme, Z.; Lewandowski, C.; Herbein, G. Distinct Oncogenic Transcriptomes in Human Mammary Epithelial Cells Infected with Cytomegalovirus. Front. Immunol. 2021, 12, 772160. [Google Scholar] [CrossRef]
- Bughio, F.; Elliott, D.A.; Goodrum, F. An Endothelial Cell-Specific Requirement for the UL133-UL138 Locus of Human Cytomegalovirus for Efficient Virus Maturation. J. Virol. 2013, 87, 3062–3075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mlera, L.; Moy, M.; Maness, K.; Tran, L.N.; Goodrum, F.D. The Role of the Human Cytomegalovirus UL133-UL138 Gene Locus in Latency and Reactivation. Viruses 2020, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Scholz, M.; Kotchetkov, R.; Vogel, J.-U.; Wilhelm Doerr, H. Molecular Mechanisms of the Modulatory Effects of HCMV Infection in Tumor Cell Biology. Trends Mol. Med. 2004, 10, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.W. The MHC Class I Antigen Presentation Pathway: Strategies for Viral Immune Evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef]
- Gilbert, M.J.; Riddell, S.R.; Li, C.R.; Greenberg, P.D. Selective Interference with Class I Major Histocompatibility Complex Presentation of the Major Immediate-Early Protein Following Infection with Human Cytomegalovirus. J. Virol. 1993, 67, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Vossen, M.; Westerhout, E.; Söderberg-Nauclér, C.; Wiertz, E. Viral Immune Evasion: A Masterpiece of Evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Vlahava, V.-M.; Forbes, S.K.; Fielding, C.A.; Stanton, R.J.; Wang, E.C.Y. HCMV-Encoded NK Modulators: Lessons from in Vitro and in Vivo Genetic Variation. Front. Immunol. 2018, 9, 2214. [Google Scholar] [CrossRef]
- Sun, J.; Lanier, L. The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors. Viruses 2009, 1, 362. [Google Scholar] [CrossRef]
- Ulbrecht, M.; Martinozzi, S.; Grzeschik, M.; Hengel, H.; Ellwart, J.W.; Pla, M.; Weiss, E.H. Cutting Edge: The Human Cytomegalovirus UL40 Gene Product Contains a Ligand for HLA-E and Prevents NK Cell-Mediated Lysis. J. Immunol. 2000, 164, 5019–5022. [Google Scholar] [CrossRef]
- Tomasec, P. Surface Expression of HLA-E, an Inhibitor of Natural Killer Cells, Enhanced by Human Cytomegalovirus GpUL40. Science 2000, 287, 1031–1033. [Google Scholar] [CrossRef]
- Spencer, J.V.; Lockridge, K.M.; Barry, P.A.; Lin, G.; Tsang, M.; Penfold, M.E.T.; Schall, T.J. Potent Immunosuppressive Activities of Cytomegalovirus- Encoded Interleukin-10. J. Virol. 2002, 76, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Crough, T.; Khanna, R. Immunobiology of Human Cytomegalovirus: From Bench to Bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Aicheler, R.; Stanton, R.J.; Wang, E.C.Y.; Han, S.; Seirafian, S.; Davies, J.; McSharry, B.P.; Weekes, M.P.; Antrobus, P.R.; et al. Two Novel Human Cytomegalovirus NK Cell Evasion Functions Target MICA for Lysosomal Degradation. PLoS Pathog 2014, 10, e1004058. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host Immune System Gene Targeting by a Viral MiRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef]
- Mumprecht, S.; Schürch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed Death 1 Signaling on Chronic Myeloid Leukemia-Specific T Cells Results in T-Cell Exhaustion and Disease Progression. Blood 2009, 114, 1528–1536. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Tovar-Salazar, A.; Weinberg, A. Understanding the Mechanism of Action of Cytomegalovirus-Induced Regulatory T Cells. Virology 2020, 547, 1–6. [Google Scholar] [CrossRef]
- Dumitriu, I.E. The Life (and Death) of CD4+ CD28(Null) T Cells in Inflammatory Diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef]
- Broadley, I.; Pera, A.; Morrow, G.; Davies, K.A.; Kern, F. Expansions of Cytotoxic CD4+CD28− T Cells Drive Excess Cardiovascular Mortality in Rheumatoid Arthritis and Other Chronic Inflammatory Conditions and Are Triggered by CMV Infection. Front. Immunol. 2017, 8, 195. [Google Scholar] [CrossRef]
- Li, M.; Boddeda, S.R.; Chen, B.; Zeng, Q.; Schoeb, T.R.; Velazquez, V.M.; Shimamura, M. NK Cell and Th17 Responses Are Differentially Induced in Murine Cytomegalovirus Infected Renal Allografts and Vary According to Recipient Virus Dose and Strain. Am. J. Transplant. 2018, 18, 2647–2662. [Google Scholar] [CrossRef]
- Curtis, M.M.; Way, S.S. Interleukin-17 in Host Defence against Bacterial, Mycobacterial and Fungal Pathogens. Immunology 2009, 126, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Agalioti, T.; Zhao, J.; Steglich, B.; Wahib, R.; Vesely, M.C.A.; Bielecki, P.; Bailis, W.; Jackson, R.; Perez, D.; et al. The Induction and Function of the Anti-Inflammatory Fate of TH17 Cells. Nat. Commun. 2020, 11, 3334. [Google Scholar] [CrossRef]
- Han, S.; Toker, A.; Liu, Z.Q.; Ohashi, P.S. Turning the Tide Against Regulatory T Cells. Front. Oncol. 2019, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.-H.; Yang, Y.-P.; Lin, J.-C.; Hsu, C.-H.; Jhang, H.-C.; Yang, Y.-T.; Lee, C.-H.; Ho, L.L.T.; Hsu, W.-M.; Ku, H.-H.; et al. The Immediate Early 2 Protein of Human Cytomegalovirus (HCMV) Mediates the Apoptotic Control in HCMV Retinitis through Up-Regulation of the Cellular FLICE-Inhibitory Protein Expression. J. Immunol. 2006, 177, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Malipiero, U.; Aguzzi, A.; Reed, J.C.; Fontana, A. Protooncogene Bcl-2 Gene Transfer Abrogates Fas/APO-1 Antibody-Mediated Apoptosis of Human Malignant Glioma Cells and Confers Resistance to Chemotherapeutic Drugs and Therapeutic Irradiation. J. Clin. Investig. 1995, 95, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Morgan, B.P.; Tufaro, F.; Devine, D.V. Altered Expression of Host-Encoded Complement Regulators on Human Cytomegalovirus-Infected Cells. Eur. J. Immunol. 1996, 26, 1532–1538. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 Gene Product of Human Cytomegalovirus Activates NFkappaB via a TRAF6-Dependent Mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef]
- La Rosa, C.; Diamond, D.J. The Immune Response to Human CMV. Future Virol. 2012, 7, 279–293. [Google Scholar] [CrossRef]
- Lepiller, Q.; Abbas, W.; Kumar, A.; Tripathy, M.K.; Herbein, G. HCMV Activates the IL-6-JAK-STAT3 Axis in HepG2 Cells and Primary Human Hepatocytes. PLoS ONE 2013, 8, e59591. [Google Scholar] [CrossRef]
- Kumar, A.; Tripathy, M.K.; Pasquereau, S.; Al Moussawi, F.; Abbas, W.; Coquard, L.; Khan, K.A.; Russo, L.; Algros, M.-P.; Valmary-Degano, S.; et al. The Human Cytomegalovirus Strain DB Activates Oncogenic Pathways in Mammary Epithelial Cells. EBioMedicine 2018, 30, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Szulc-Kielbik, I.; Kielbik, M.; Nowak, M.; Klink, M. The Implication of IL-6 in the Invasiveness and Chemoresistance of Ovarian Cancer Cells. Systematic Review of Its Potential Role as a Biomarker in Ovarian Cancer Patients. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188639. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Human Cytomegalovirus Stimulates Monocyte-to-Macrophage Differentiation via the Temporal Regulation of Caspase 3. J. Virol. 2012, 86, 10714–10723. [Google Scholar] [CrossRef] [PubMed]
- Cojohari, O.; Mahmud, J.; Altman, A.M.; Peppenelli, M.A.; Miller, M.J.; Chan, G.C. Human Cytomegalovirus Mediates Unique Monocyte-to-Macrophage Differentiation through the PI3K/SHIP1/Akt Signaling Network. Viruses 2020, 12, 652. [Google Scholar] [CrossRef] [PubMed]
- Bayer, C.; Varani, S.; Wang, L.; Walther, P.; Zhou, S.; Straschewski, S.; Bachem, M.; Söderberg-Naucler, C.; Mertens, T.; Frascaroli, G. Human Cytomegalovirus Infection of M1 and M2 Macrophages Triggers Inflammation and Autologous T-Cell Proliferation. J. Virol. 2013, 87, 67–79. [Google Scholar] [CrossRef]
- Khan, K.A.; Coaquette, A.; Davrinche, C.; Herbein, G. Bcl-3-Regulated Transcription from Major Immediate-Early Promoter of Human Cytomegalovirus in Monocyte-Derived Macrophages. J. Immunol. 2009, 182, 7784–7794. [Google Scholar] [CrossRef]
- Margulies, B.J.; Browne, H.; Gibson, W. Identification of the Human Cytomegalovirus G Protein-Coupled Receptor Homologue Encoded by UL33 in Infected Cells and Enveloped Virus Particles. Virology 1996, 225, 111–125. [Google Scholar] [CrossRef]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-Encoded Chemokine Receptor US28 Mediates Proliferative Signaling through the IL-6-STAT3 Axis. Sci. Signal. 2010, 3, ra58. [Google Scholar] [CrossRef]
- Jackson, S.E.; Redeker, A.; Arens, R.; van Baarle, D.; van den Berg, S.P.H.; Benedict, C.A.; Čičin-Šain, L.; Hill, A.B.; Wills, M.R. CMV Immune Evasion and Manipulation of the Immune System with Aging. GeroScience 2017, 39, 273–291. [Google Scholar] [CrossRef]
- Beisser, P.S.; Laurent, L.; Virelizier, J.-L.; Michelson, S. Human Cytomegalovirus Chemokine Receptor Gene US28 Is Transcribed in Latently Infected THP-1 Monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef]
- Young, V.P.; Mariano, M.C.; Tu, C.C.; Allaire, K.M.; Avdic, S.; Slobedman, B.; Spencer, J.V. Modulation of the Host Environment by Human Cytomegalovirus with Viral Interleukin 10 in Peripheral Blood. J. Infect. Dis. 2017, 215, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Michelson, S.; Alcami, J.; Kim, S.J.; Danielpour, D.; Bachelerie, F.; Picard, L.; Bessia, C.; Paya, C.; Virelizier, J.L. Human Cytomegalovirus Infection Induces Transcription and Secretion of Transforming Growth Factor Beta 1. J. Virol. 1994, 68, 5730–5737. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Baasch, S.; Giansanti, P.; Kolter, J.; Riedl, A.; Forde, A.J.; Runge, S.; Zenke, S.; Elling, R.; Halenius, A.; Brabletz, S.; et al. Cytomegalovirus Subverts Macrophage Identity. Cell 2021, 184, 3774–3793.e25. [Google Scholar] [CrossRef] [PubMed]
- Caposio, P.; Orloff, S.L.; Streblow, D.N. The Role of Cytomegalovirus in Angiogenesis. Virus Res. 2011, 157, 204–211. [Google Scholar] [CrossRef]
- Reitsma, J.M.; Sato, H.; Nevels, M.; Terhune, S.S.; Paulus, C. Human Cytomegalovirus IE1 Protein Disrupts Interleukin-6 Signaling by Sequestering STAT3 in the Nucleus. J. Virol. 2013, 87, 10763–10776. [Google Scholar] [CrossRef]
- Valle Oseguera, C.A.; Spencer, J.V. CmvIL-10 Stimulates the Invasive Potential of MDA-MB-231 Breast Cancer Cells. PLoS ONE 2014, 9, e88708. [Google Scholar] [CrossRef]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating Neutrophils Mediate the Initial Angiogenic Switch in a Mouse Model of Multistage Carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef]
- Rahbar, A.; Cederarv, M.; Wolmer-Solberg, N.; Tammik, C.; Stragliotto, G.; Peredo, I.; Fornara, O.; Xu, X.; Dzabic, M.; Taher, C.; et al. Enhanced Neutrophil Activity Is Associated with Shorter Time to Tumor Progression in Glioblastoma Patients. OncoImmunology 2016, 5, e1075693. [Google Scholar] [CrossRef]
- Teo, W.H.; Chen, H.-P.; Huang, J.C.; Chan, Y.-J. Human Cytomegalovirus Infection Enhances Cell Proliferation, Migration and Upregulation of EMT Markers in Colorectal Cancer-Derived Stem Cell-like Cells. Int. J. Oncol. 2017, 51, 1415–1426. [Google Scholar] [CrossRef]
- Shimamura, M.; Murphy-Ullrich, J.E.; Britt, W.J. Human Cytomegalovirus Induces TGF-Β1 Activation in Renal Tubular Epithelial Cells after Epithelial-to-Mesenchymal Transition. PLoS Pathog 2010, 6, e1001170. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, M.; Paggi, M.G.; Giordano, A. Genetic and Epigenetic Alterations as Hallmarks of the Intricate Road to Cancer. Oncogene 2003, 22, 6472–6478. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C. Cytomegalovirus Is a Tumor-Associated Virus: Armed and Dangerous. Curr. Opin. Virol. 2019, 39, 49–59. [Google Scholar] [CrossRef]
- Taher, C.; de Boniface, J.; Mohammad, A.-A.; Religa, P.; Hartman, J.; Yaiw, K.-C.; Frisell, J.; Rahbar, A.; Söderberg-Naucler, C. High Prevalence of Human Cytomegalovirus Proteins and Nucleic Acids in Primary Breast Cancer and Metastatic Sentinel Lymph Nodes. PLoS ONE 2013, 8, e56795. [Google Scholar] [CrossRef]
- Taher, C.; Frisk, G.; Fuentes, S.; Religa, P.; Costa, H.; Assinger, A.; Vetvik, K.K.; Bukholm, I.R.K.; Yaiw, K.-C.; Smedby, K.E.; et al. High Prevalence of Human Cytomegalovirus in Brain Metastases of Patients with Primary Breast and Colorectal Cancers. Transl. Oncol. 2014, 7, 732–740. [Google Scholar] [CrossRef]
- Harkins, L.E.; Matlaf, L.A.; Soroceanu, L.; Klemm, K.; Britt, W.J.; Wang, W.; Bland, K.I.; Cobbs, C.S. Detection of Human Cytomegalovirus in Normal and Neoplastic Breast Epithelium. Herpesviridae 2010, 1, 8. [Google Scholar] [CrossRef]
- Rahbar, A.; Touma, J.; Costa, H.; Davoudi, B.; Bukholm, I.R.; Sauer, T.; Vetvik, K.; Geisler, J.; Söderberg-Naucler, C. Low Expression of Estrogen Receptor-α and Progesterone Receptor in Human Breast Cancer Tissues Is Associated with High-Grade Human Cytomegalovirus Protein Expression. Clin. Breast Cancer 2017, 17, 526–535.e1. [Google Scholar] [CrossRef]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsum, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The Human Cytomegalovirus-Encoded Chemokine Receptor US28 Promotes Angiogenesis and Tumor Formation via Cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; Bland, K.I.; Cobbs, C.S. Specific Localisation of Human Cytomegalovirus Nucleic Acids and Proteins in Human Colorectal Cancer. The Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe, J.; Alshafie, G.A. Reduction in Cancer Risk by Selective and Nonselective Cyclooxygenase-2 (COX-2) Inhibitors. J. Exp. Pharmacol. 2012, 4, 91–96. [Google Scholar] [CrossRef]
- Samanta, M.; Harkins, L.; Klemm, K.; Britt, W.J.; Cobbs, C.S. High Prevalence of Human Cytomegalovirus in Prostatic Intraepithelial Neoplasia and Prostatic Carcinoma. J. Urol. 2003, 170, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Wolmer-Solberg, N.; Baryawno, N.; Rahbar, A.; Fuchs, D.; Odeberg, J.; Taher, C.; Wilhelmi, V.; Milosevic, J.; Mohammad, A.-A.; Martinsson, T.; et al. Frequent Detection of Human Cytomegalovirus in Neuroblastoma: A Novel Therapeutic Target?: Human Cytomegalovirus in Neuroblastoma. Int. J. Cancer 2013, 133, 2351–2361. [Google Scholar] [CrossRef]
- Baryawno, N.; Rahbar, A.; Wolmer-Solberg, N.; Taher, C.; Odeberg, J.; Darabi, A.; Khan, Z.; Sveinbjörnsson, B.; FuskevÅg, O.-M.; Segerström, L.; et al. Detection of Human Cytomegalovirus in Medulloblastomas Reveals a Potential Therapeutic Target. J. Clin. Investig. 2011, 121, 4043–4055. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wilkie, A.R.; Weller, M.; Liu, X.; Cohen, J.I. THY-1 Cell Surface Antigen (CD90) Has an Important Role in the Initial Stage of Human Cytomegalovirus Infection. PLoS Pathog 2015, 11, e1004999. [Google Scholar] [CrossRef]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-Derived Growth Factor-α Receptor Activation Is Required for Human Cytomegalovirus Infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef]
- Stegmann, C.; Hochdorfer, D.; Lieber, D.; Subramanian, N.; Stöhr, D.; Laib Sampaio, K.; Sinzger, C. A Derivative of Platelet-Derived Growth Factor Receptor Alpha Binds to the Trimer of Human Cytomegalovirus and Inhibits Entry into Fibroblasts and Endothelial Cells. PLoS Pathog 2017, 13, e1006273. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human Cytomegalovirus Glycoprotein Complex GH/GL/GO Uses PDGFR-α as a Key for Entry. PLoS Pathog 2017, 13, e1006281. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-W.; Yang, D.; Yang, D.; Chen, Z.; Miao, J.; Liu, W.; Wang, X.; Qiu, Z.; Jin, M.; Shen, Z. Tumors Arise from the Excessive Repair of Damaged Stem Cells. Med. Hypotheses 2017, 102, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or Abusing: Viruses and the Cellular DNA Damage Response. Trends Microbiol. 2007, 15, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Matlaf, L.; Khan, S.; Akhavan, A.; Singer, E.; Bezrookove, V.; Decker, S.; Ghanny, S.; Hadaczek, P.; Bengtsson, H.; et al. Cytomegalovirus Immediate-Early Proteins Promote Stemness Properties in Glioblastoma. Cancer Res. 2015, 75, 3065–3076. [Google Scholar] [CrossRef]
- Fornara, O.; Bartek, J.; Rahbar, A.; Odeberg, J.; Khan, Z.; Peredo, I.; Hamerlik, P.; Bartek, J.; Stragliotto, G.; Landázuri, N.; et al. Cytomegalovirus Infection Induces a Stem Cell Phenotype in Human Primary Glioblastoma Cells: Prognostic Significance and Biological Impact. Cell Death Differ. 2016, 23, 261–269. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Soroceanu, L.; Denham, S.; Zhang, W.; Kraus, M.H. Modulation of Oncogenic Phenotype in Human Glioma Cells by Cytomegalovirus IE1–Mediated Mitogenicity. Cancer Res. 2008, 68, 724–730. [Google Scholar] [CrossRef]
- Haidar Ahmad, S.; Al Moussawi, F.; El Baba, R.; Nehme, Z.; Pasquereau, S.; Kumar, A.; Molimard, C.; Monnien, F.; Algros, M.-P.; Karaky, R.; et al. Identification of UL69 Gene and Protein in Cytomegalovirus-Transformed Human Mammary Epithelial Cells. Front. Oncol. 2021, 11, 627866. [Google Scholar] [CrossRef]
- Moussawi, F.A.; Kumar, A.; Pasquereau, S.; Tripathy, M.K.; Karam, W.; Diab-Assaf, M.; Herbein, G. The Transcriptome of Human Mammary Epithelial Cells Infected with the HCMV-DB Strain Displays Oncogenic Traits. Sci. Rep. 2018, 8, 12574. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Spector, D.H. Viral Induction of Site-Specific Chromosome Damage. Rev. Med. Virol. 2003, 13, 21–37. [Google Scholar] [CrossRef]
- Siew, V.-K.; Duh, C.-Y.; Wang, S.-K. Human Cytomegalovirus UL76 Induces Chromosome Aberrations. J. Biomed. Sci. 2009, 16, 107. [Google Scholar] [CrossRef]
- Cojohari, O.; Peppenelli, M.A.; Chan, G.C. Human Cytomegalovirus Induces an Atypical Activation of Akt To Stimulate the Survival of Short-Lived Monocytes. J. Virol. 2016, 90, 6443–6452. [Google Scholar] [CrossRef] [PubMed]
- Altman, A.M.; Mahmud, J.; Nikolovska-Coleska, Z.; Chan, G. HCMV Modulation of Cellular PI3K/AKT/MTOR Signaling: New Opportunities for Therapeutic Intervention? Antiviral. Res. 2019, 163, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Baylot, V.; Felsher, D.W. MYC: Master Regulator of Immune Privilege. Trends Immunol. 2017, 38, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Villegas, S.N.; Gombos, R.; García-López, L.; Gutiérrez-Pérez, I.; García-Castillo, J.; Vallejo, D.M.; Da Ros, V.G.; Ballesta-Illán, E.; Mihály, J.; Dominguez, M. PI3K/Akt Cooperates with Oncogenic Notch by Inducing Nitric Oxide-Dependent Inflammation. Cell Rep. 2018, 22, 2541–2549. [Google Scholar] [CrossRef]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef]
- Niu, N.; Yao, J.; Bast, R.C.; Sood, A.K.; Liu, J. IL-6 Promotes Drug Resistance through Formation of Polyploid Giant Cancer Cells and Stromal Fibroblast Reprogramming. Oncogenesis 2021, 10, 65. [Google Scholar] [CrossRef]
- Stragliotto, G.; Rahbar, A.; Solberg, N.W.; Lilja, A.; Taher, C.; Orrego, A.; Bjurman, B.; Tammik, C.; Skarman, P.; Peredo, I.; et al. Effects of Valganciclovir as an Add-on Therapy in Patients with Cytomegalovirus-Positive Glioblastoma: A Randomized, Double-Blind, Hypothesis-Generating Study. Int. J. Cancer 2013, 133, 1204–1213. [Google Scholar] [CrossRef]
- Stragliotto, G.; Pantalone, M.R.; Rahbar, A.; Bartek, J.; Söderberg-Naucler, C. Valganciclovir as Add-on to Standard Therapy in Glioblastoma Patients. Clin. Cancer Res. 2020, 26, 4031–4039. [Google Scholar] [CrossRef]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Weathers, S.-P.; Penes-Prado, M.; Pei, B.-L.; Ling, X.; Kassab, C.; Banerjee, P.; Bdiwi, M.; Shaim, H.; Al-suliman, A.; Shanley, M.; et al. Glioblastoma-mediated immune dysfunction limits CMV-specific T cells and therapeutic responses: Results from a phase I/II trial. Clin. Cancer. Res. 2020, 26, 3565–3577. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; Erkes, D.A.; Snyder, C.M. Cytomegalovirus and Immunotherapy: Opportunistic Pathogen, Novel Target for Cancer and a Promising Vaccine Vector. Immunotherapy 2016, 8, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Qiu, Y.; Zhang, Z.; Wu, M. Insight for Immunotherapy of HCMV Infection. Int. J. Biol. Sci. 2021, 17, 2899–2911. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbein, G. Tumors and Cytomegalovirus: An Intimate Interplay. Viruses 2022, 14, 812. https://doi.org/10.3390/v14040812
Herbein G. Tumors and Cytomegalovirus: An Intimate Interplay. Viruses. 2022; 14(4):812. https://doi.org/10.3390/v14040812
Chicago/Turabian StyleHerbein, Georges. 2022. "Tumors and Cytomegalovirus: An Intimate Interplay" Viruses 14, no. 4: 812. https://doi.org/10.3390/v14040812
APA StyleHerbein, G. (2022). Tumors and Cytomegalovirus: An Intimate Interplay. Viruses, 14(4), 812. https://doi.org/10.3390/v14040812