
Target Enrichment Metagenomics Reveals Human Pegivirus-1 in Pediatric Hematopoietic Stem Cell Transplantation Recipients

 , , , , and
, , , , and
Abstract
1. Introduction
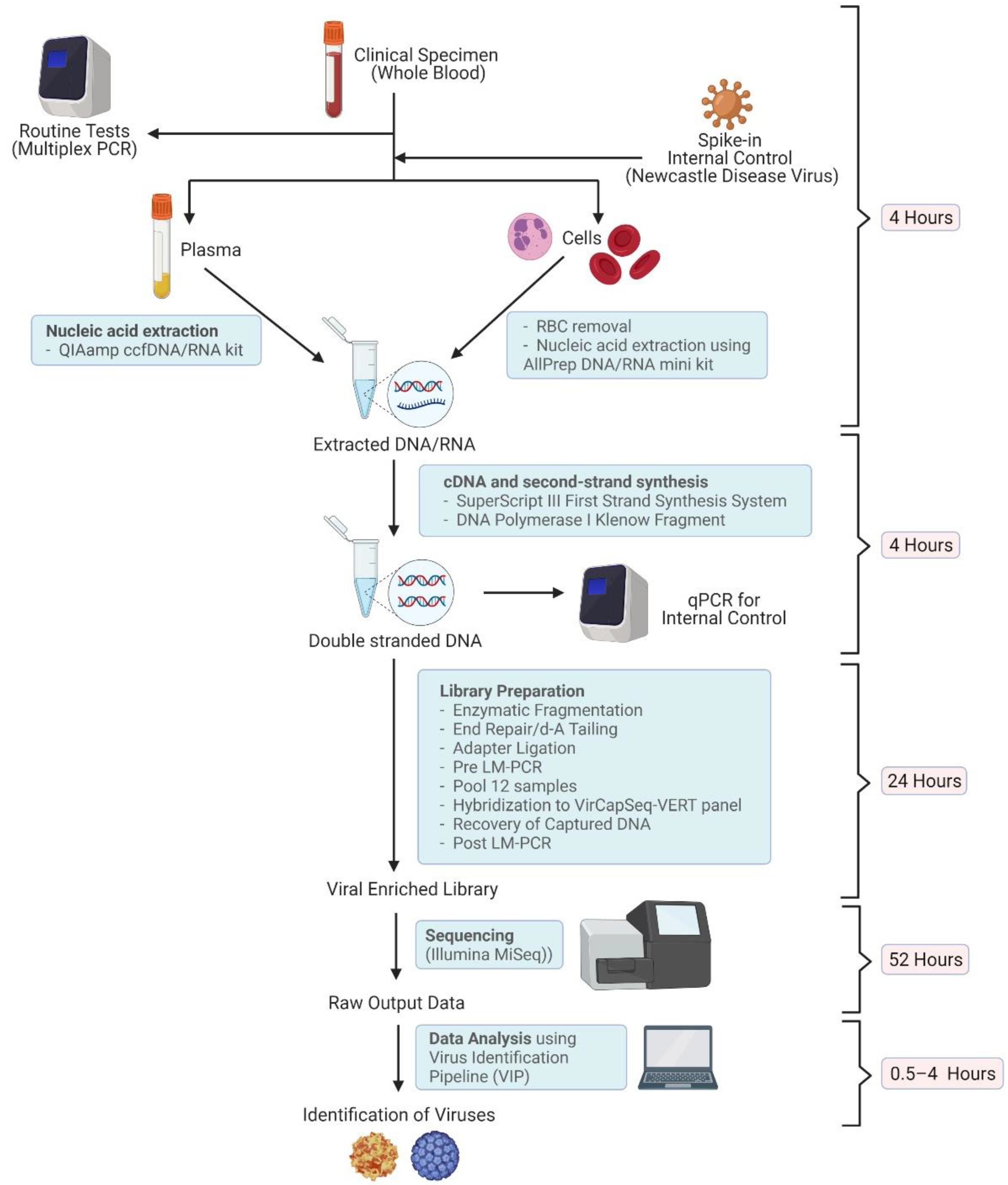
2. Methods
2.1. Patients and Samples
2.2. Nucleic Acid Extraction
2.3. Library Preparation
2.4. Sequencing
2.5. Virus Identification
2.6. Genome Assembly
2.7. Phylogenetic Reconstruction
3. Results
3.1. Enrolled Patients
3.2. Virus Detection by Routine Tests
3.3. Virus Detection by TE-NGS Technology
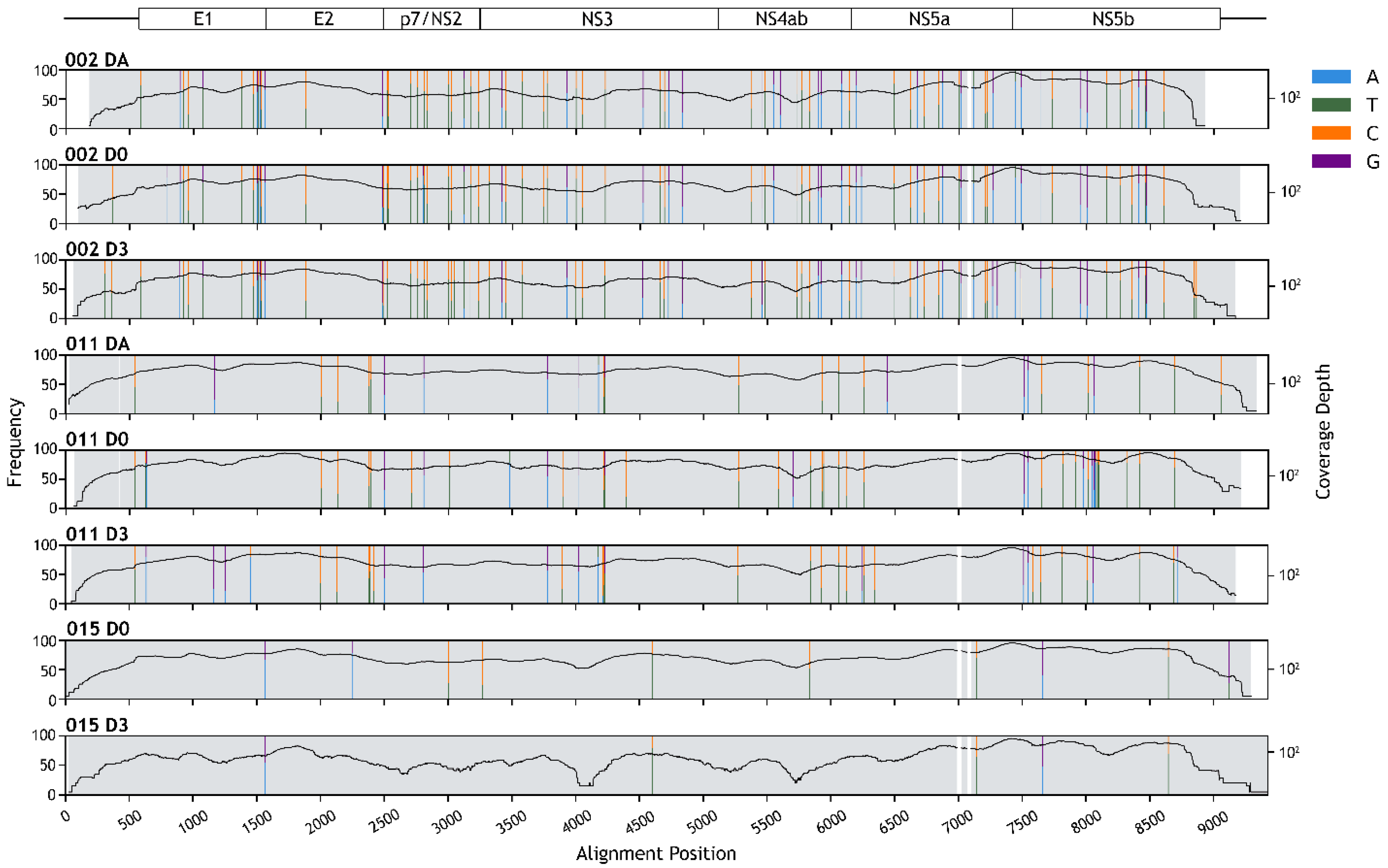
3.4. HPgV-1 Genome Assembly
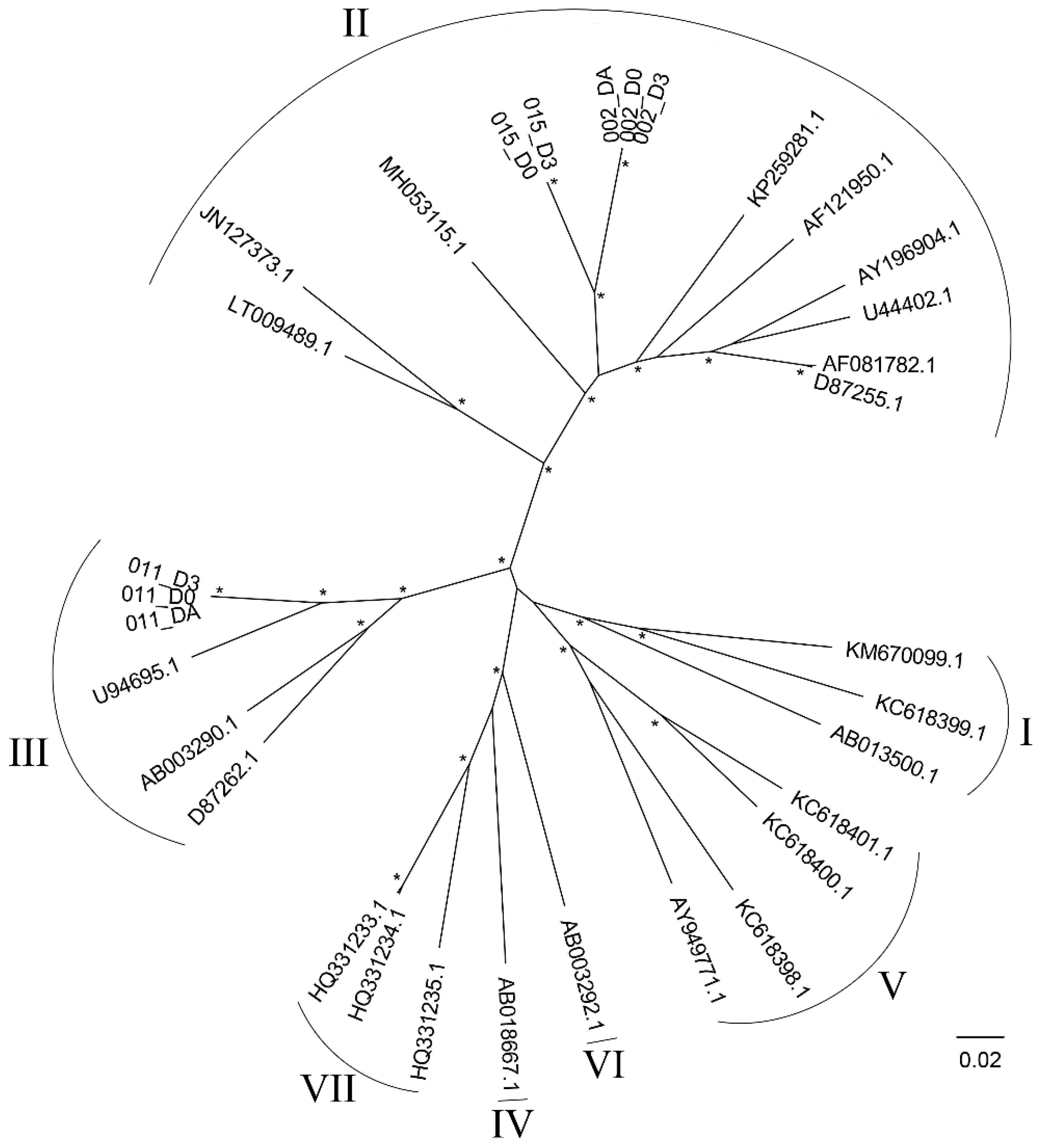
3.5. Genotyping
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mrzljak, A.; Simunov, B.; Balen, I.; Jurekovic, Z.; Vilibic-Cavlek, T. Human pegivirus infection after transplant: Is there an impact? World J. Transplant. 2022, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, J.T. GB Virus Type C/Hepatitis G Virus In Seminars in Liver Disease; Thieme Medical Publishers, Inc.: New York, NY, USA, 2003; pp. 584–4662. [Google Scholar]
- Izumi, T.; Sakata, K.; Okuzaki, D.; Inokuchi, S.; Tamura, T.; Motooka, D.; Nakamura, S.; Ono, C.; Shimokawa, M.; Matsuura, Y.; et al. Characterization of human pegivirus infection in liver transplantation recipients. J. Med. Virol. 2019, 91, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Sathar, M.; Soni, P.; York, D. GB virus C/hepatitis G virus (GBV-C/HGV): Still looking for a disease. Int. J. Exp. Pathol. 2000, 81, 305–322. [Google Scholar] [CrossRef]
- Ramos Filho, R.; Carneiro, M.A.; Teles, S.A.; Dias, M.A.; Cardoso, D.D.; Lampe, E.; Yoshida, C.F.; Martins, R. GB virus C/hepatitis G virus infection in dialysis patients and kidney transplant recipients in Central Brazil. Mem. Inst. Oswaldo Cruz 2004, 99, 639–643. [Google Scholar] [CrossRef]
- Abraham, P.; John, G.T.; Raghuraman, S.; Radhakrishnan, S.; Thomas, P.P.; Jacob, C.K.; Sridharan, G. GB virus C/hepatitis G virus and TT virus infections among high risk renal transplant recipients in India. J. Clin. Virol. 2003, 28, 59–69. [Google Scholar] [CrossRef]
- Kalkan, A.; Ozdarendeli, A.; Bulut, Y.; Saral, Y.; Ozden, M.; Kelestimur, N.; Toraman, Z.A. Prevalence and genotypic distribution of hepatitis GB-C/HG and TT viruses in blood donors, mentally retarded children and four groups of patients in eastern Anatolia, Turkey. Jpn. J. Infect. Dis. 2005, 58, 222. [Google Scholar]
- Slavov, S.N.; Silveira, R.M.; Rodrigues, E.S.; Diefenbach, C.F.; Zimmermann, A.M.; Covas, D.T.; Kashima, S. Human pegivirus-1 (HPgV-1, GBV-C) RNA prevalence and genotype diversity among volunteer blood donors from an intra-hospital hemotherapy service in Southern Brazil. Transfus. Apher. Sci. 2019, 58, 174–178. [Google Scholar] [CrossRef]
- Tuddenham, R.; Eden, J.S.; Gilbey, T.; Dwyer, D.E.; Jennings, Z.; Holmes, E.C.; Branley, J.M. Human pegivirus in brain tissue of a patient with encephalitis. Diagn. Microbiol. Infect. Dis. 2020, 96, 114898. [Google Scholar] [CrossRef]
- Raengsakulrach, B.; Ong-aj-yooth, L.; Thaiprasert, T.; Nilwarangkur, S.; Ong-aj-yooth, S.; Narupiti, S.; Thirawuth, V.; Klungthong, C.; Snitbhan, R.; Vaughn, D.W. High prevalence of hepatitis G viremia among kidney transplant patients in Thailand. J. Med. Virol. 1997, 53, 162–166. [Google Scholar] [CrossRef]
- Poovorawan, Y.; Theamboonlers, A.; Chongsrisawat, V.; Jantaradsamee, P. Prevalence of infection with hepatitis G virus among various groups in Thailand. Ann. Trop. Med. Parasitol. 1998, 92, 89–95. [Google Scholar] [CrossRef]
- Bhanich Supapol, W.; Remis, R.; Raboud, J.; Millson, M.; Tappero, J.; Kaul, R.; Kulkarni, P.; McConnell, M.; Mock, P.; McNicholl, J. Mother-to-child transmission of GB virus C in a cohort of women coinfected with GB virus C and HIV in Bangkok, Thailand. J. Infect. Dis. 2009, 200, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Stäuber, N.; Brechtbühl, K.; Bruckner, L.; Hofmann, M.A. Detection of Newcastle disease virus in poultry vaccines using the polymerase chain reaction and direct sequencing of amplified cDNA. Vaccine 1995, 13, 360–364. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An integrated pipeline for metagenomics of virus identification and discovery. Sci. Rep. 2016, 6, 23774. [Google Scholar] [CrossRef]
- Shahzamani, K.; Jahanbakhsh, S.; Lashgarian, H. Qualitative detection of GB Virus C and Hepatitis C Virus co-infection in cirrhotic patients using a SYBR green multiplex RT-PCR technique. Trop. Biomed. 2017, 34, 822–830. [Google Scholar] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Chakraborty, M.; Baldwin-Brown, J.G.; Long, A.D.; Emerson, J.J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016, 44, e147. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Bukowska-Ośko, I.; Perlejewski, K.; Pawełczyk, A.; Rydzanicz, M.; Pollak, A.; Popiel, M.; Cortés, K.C.; Paciorek, M.; Horban, A.; Dzieciątkowski, T. Human Pegivirus in patients with encephalitis of unclear etiology, Poland. Emerg. Infect. Dis. 2018, 24, 1785. [Google Scholar] [CrossRef]
- Baldwin, A.; Kingman, H.; Darville, M.; Foot, A.; Grier, D.; Cornish, J.; Goulden, N.; Oakhill, A.; Pamphilon, D.; Steward, C. Outcome and clinical course of 100 patients with adenovirus infection following bone marrow transplantation. Bone Marrow Transplant. 2000, 26, 1333–1338. [Google Scholar] [CrossRef]
- George, B.; Mathews, V.; Srivastava, A.; Chandy, M. Infections among allogeneic bone marrow transplant recipients in India. Bone Marrow Transplant. 2004, 33, 311–315. [Google Scholar] [CrossRef][Green Version]
- Ljungman, P. Molecular monitoring of viral infections after hematopoietic stem cell transplantation. Int. J. Hematol. 2010, 91, 596–601. [Google Scholar] [CrossRef]
- Düver, F.; Weißbrich, B.; Eyrich, M.; Wölfl, M.; Schlegel, P.G.; Wiegering, V. Viral reactivations following hematopoietic stem cell transplantation in pediatric patients–A single center 11-year analysis. PLoS ONE 2020, 15, e0228451. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, Q. Diagnosis and treatment of viral diseases in recipients of allogeneic hematopoietic stem cell transplantation. J. Hematol. 2013, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Kaeuferle, T.; Krauss, R.; Blaeschke, F.; Willier, S.; Feuchtinger, T. Strategies of adoptive T-cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Breuer, S.; Rauch, M.; Matthes-Martin, S.; Lion, T. Molecular diagnosis and management of viral infections in hematopoietic stem cell transplant recipients. Mol. Diagn. Ther. 2012, 16, 63–77. [Google Scholar] [CrossRef]
- Alexandersson, A.; Koskenvuo, M.; Tiderman, A.; Lääperi, M.; Huttunen, P.; Saarinen-Pihkala, U.; Anttila, V.-J.; Lautenschlager, I.; Taskinen, M. Viral infections and immune reconstitution interaction after pediatric allogenic hematopoietic stem cell transplantation. J. Infect. Dis. 2019, 51, 772–778. [Google Scholar] [CrossRef]
- Zanella, M.C.; Cordey, S.; Laubscher, F.; Docquier, M.; Vieille, G.; Van Delden, C.; Braunersreuther, V.; Ta, M.K.; Lobrinus, J.A.; Masouridi-Levrat, S.; et al. Unmasking viral sequences by metagenomic next-generation sequencing in adult human blood samples during steroid-refractory/dependent graft-versus-host disease. Microbiome 2021, 9, 28. [Google Scholar] [CrossRef]
- Datta, S.; Budhauliya, R.; Das, B.; Chatterjee, S. Next-generation sequencing in clinical virology: Discovery of new viruses. World J. Virol. 2015, 4, 265. [Google Scholar] [CrossRef]
- Zárate, S.; Taboada, B.; Yocupicio-Monroy, M.; Arias, C.F. Human virome. Arch. Med. Res. 2017, 48, 701–716. [Google Scholar] [CrossRef]
- Lecuit, M.; Eloit, M. The human virome: New tools and concepts. Trends Microbiol. 2013, 21, 510–515. [Google Scholar] [CrossRef]
- Chalkias, S.; Gorham, J.M.; Mazaika, E.; Parfenov, M.; Dang, X.; DePalma, S.; McKean, D.; Seidman, C.E.; Seidman, J.G.; Koralnik, I.J. ViroFind: A novel target-enrichment deep-sequencing platform reveals a complex JC virus population in the brain of PML patients. PLoS ONE 2018, 13, 0186945. [Google Scholar] [CrossRef]
- Jensen, R.H.; Mollerup, S.; Mourier, T.; Hansen, T.A.; Fridholm, H.; Nielsen, L.P.; Willerslev, E.; Hansen, A.J.; Vinner, L. Target-dependent enrichment of virions determines the reduction of high-throughput sequencing in virus discovery. PLoS ONE 2015, 10, 0122636. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.D.; Bui, V.N.; Omatsu, T.; Katayama, Y.; Mizutani, T.; Ogawa, H.; Imai, K. Application of the SureSelect target enrichment system for next-generation sequencing to obtain the complete genome sequence of bovine leukemia virus. Arch. Virol. 2018, 163, 3155–3159. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, N.; Kobayashi, T.; Matsuo, M.; Nishikaku, K.; Kusama, K.; Torii, Y.; Inagaki, Y.; Hori, M.; Imakawa, K.; Satou, Y. A target enrichment high throughput sequencing system for characterization of BLV whole genome sequence, integration sites, clonality and host SNP. Sci. Rep. 2021, 11, 4521. [Google Scholar] [CrossRef] [PubMed]
- McGill, F.; Tokarz, R.; Thomson, E.C.; Filipe, A.; Sameroff, S.; Jain, K.; Bhuva, N.; Ashraf, S.; Lipkin, W.I.; Corless, C. Viral capture sequencing detects unexpected viruses in the cerebrospinal fluid of adults with meningitis. J. Infect. 2022; in press. [Google Scholar] [CrossRef]
- Tan, M.T.H.; Ho, S.X.; Chu, J.J.H.; Li, D. Application of virome capture sequencing in shellfish sold at retail level in Singapore. Lett. Appl. Microbiol. 2021, 73, 486–494. [Google Scholar] [CrossRef]
- Ho, S.X.; Min, N.; Wong, E.P.Y.; Chong, C.Y.; Chu, J.J.H. Characterization of oral virome and microbiome revealed distinctive microbiome disruptions in paediatric patients with hand, foot and mouth disease. NPJ Biofilms Microbiomes 2021, 7, 19. [Google Scholar] [CrossRef]
- Martínez-Puchol, S.; Rusiñol, M.; Fernández-Cassi, X.; Timoneda, N.; Itarte, M.; Andrés, C.; Antón, A.; Abril, J.F.; Girones, R.; Bofill-Mas, S. Characterisation of the sewage virome: Comparison of NGS tools and occurrence of significant pathogens. Sci. Total Environ. 2020, 713, 136604. [Google Scholar] [CrossRef]
- Forés, E.; Rusiñol, M.; Itarte, M.; Martínez-Puchol, S.; Calvo, M.; Bofill-Mas, S. Evaluation of a virus concentration method based on ultrafiltration and wet foam elution for studying viruses from large-volume water samples. Sci. Total Environ. 2022, 829, 154431. [Google Scholar] [CrossRef]
- Eloit, M. The diagnosis of infectious diseases by whole genome next generation sequencing: A new era is opening. Front. Cell. Infect. Microbiol. 2014, 4, 25. [Google Scholar]
- Li, Z.; Li, Y.; Liang, Y.; Hu, L.; Chen, S. Prevalence and risk factors of human pegivirus type 1 infection in hematopoietic stem cell transplantation patients. Int. J. Infect. Dis. 2019, 85, 111–113. [Google Scholar] [CrossRef]
- Vu, D.-L.; Cordey, S.; Simonetta, F.; Brito, F.; Docquier, M.; Turin, L.; Van Delden, C.; Boely, E.; Dantin, C.; Pradier, A. Human pegivirus persistence in human blood virome after allogeneic haematopoietic stem-cell transplantation. Clin. Microbiol. Infect. 2019, 25, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iñigo, E.; Tomás, J.-F.; Gómez-García de Soria, V.; Bartolomé, J.; Pinilla, I.; Amaro, M.A.-J.; Carreño, V.; Fernández-Rañada, J.-M. Hepatitis C and G virus infection and liver dysfunction after allogeneic bone marrow transplantation: Results from a prospective study. Blood J. Am. Soc. Hematol. 1997, 90, 1326–1331. [Google Scholar]
- Corbi, C.; Traineau, R.; Esperou, H.; Ravera, N.; Portelette, E.; Benbunan, M.; Gluckman, E.; Loiseau, P. Prevalence and clinical features of hepatitis G virus infection in bone marrow allograft recipients. Bone Marrow Transplant. 1997, 20, 965–968. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zimmerman, J.; Blackard, J.T. Human pegivirus type 1 infection in Asia—A review of the literature. Rev. Med. Virol. 2022, 32, e2257. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, Z.; Wang, J.-H.; Yang, X.; Zhang, C. Review of human pegivirus: Prevalence, transmission, pathogenesis, and clinical implication. Virulence 2022, 13, 324–341. [Google Scholar] [CrossRef] [PubMed]
- Muccini, C.; Crowell, T.A.; Kroon, E.; Sacdalan, C.; Ramautarsing, R.; Seekaew, P.; Phanuphak, P.; Ananworanich, J.; Colby, D.J.; Phanuphak, N. Leveraging early HIV diagnosis and treatment in Thailand to conduct HIV cure research. AIDS Res. Ther. 2019, 16, 25. [Google Scholar] [CrossRef]
- Jordier, F.; Deligny, M.L.; Barré, R.; Robert, C.; Galicher, V.; Uch, R.; Fournier, P.E.; Raoult, D.; Biagini, P. Human pegivirus isolates characterized by deep sequencing from hepatitis C virus-RNA and human immunodeficiency virus-RNA–positive blood donations, France. J. Med. Virol. 2019, 91, 38–44. [Google Scholar] [CrossRef]
- Feng, Y.; Zhao, W.; Feng, Y.; Dai, J.; Li, Z.; Zhang, X.; Liu, L.; Bai, J.; Zhang, H.; Lu, L. A novel genotype of GB virus C: Its identification and predominance among injecting drug users in Yunnan, China. PLoS ONE 2011, 6, e21151. [Google Scholar] [CrossRef]
- Katayama, Y.; Apichartpiyakul, C.; Handajani, R.; Ishido, S.; Hotta, H. GB virus C/hepatitis G virus (GBV-C/HGV) infectionin Chiang Mai, Thailand, and identification of variants on the basis of 5′-untranslated region sequences. Arch. Virol. 1997, 142, 2433–2445. [Google Scholar] [CrossRef]
- Liang, Y.; Hu, F.; Fan, H.; Li, L.; Wan, Z.; Wang, H.; Shui, J.; Zhou, Y.; Tong, Y.; Cai, W. Difference of Intrahost Dynamics of the Second Human Pegivirus and Hepatitis C Virus in HPgV-2/HCV-Coinfected Patients. Front. Cell. Infect. Microbiol. 2021, 11, 728415. [Google Scholar] [CrossRef]
- Bass, B. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. ADARs: Viruses and innate immunity. In Adenosine Deaminases Acting on RNA (ADARs) and A-to-I Editing; Springer: Berlin/Heidelberg, Germany, 2011; pp. 163–195. [Google Scholar]
- Carpenter, J.A.; Keegan, L.P.; Wilfert, L.; O’Connell, M.A.; Jiggins, F.M. Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae). BMC Genet. 2009, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Piontkivska, H.; Frederick, M.; Miyamoto, M.M.; Wayne, M.L. RNA editing by the host ADAR system affects the molecular evolution of the Zika virus. Ecol. Evol. 2017, 7, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Tomaselli, S.; Galeano, F.; Locatelli, F.; Gallo, A. ADARs and the balance game between virus infection and innate immune cell response. Curr. Issues Mol. Biol. 2015, 17, 37–52. [Google Scholar]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479, 131–145. [Google Scholar] [CrossRef]
- Perelygina, L.; Chen, M.; Suppiah, S.; Adebayo, A.; Abernathy, E.; Dorsey, M.; Bercovitch, L.; Paris, K.; White, K.P.; Krol, A. Infectious vaccine-derived rubella viruses emerge, persist, and evolve in cutaneous granulomas of children with primary immunodeficiencies. PLoS Pathog. 2019, 15, e1008080. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-mediated restriction of RNA virus replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Ratcliff, J.; Simmonds, P. Potential APOBEC-mediated RNA editing of the genomes of SARS-CoV-2 and other coronaviruses and its impact on their longer term evolution. Virology 2021, 556, 62–72. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C → U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: Causes and consequences for their short-and long-term evolutionary trajectories. Msphere 2020, 5, e00408-20. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.M.; Stapleton, J.T.; Klinzman, D.; McLinden, J.H.; Purdue, M.P.; Katki, H.A.; Engels, E.A. GBV-C infection and risk of NHL among US adults. Cancer Res. 2014, 74, 5553–5560. [Google Scholar] [CrossRef] [PubMed]
- Krajden, M.; Yu, A.; Braybrook, H.; Lai, A.S.; Mak, A.; Chow, R.; Cook, D.; Tellier, R.; Petric, M.; Gascoyne, R.D. GBV-C/hepatitis G virus infection and non—Hodgkin lymphoma: A case control study. Int. J. Cancer 2010, 126, 2885–2892. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample Code | Gender | Age | Peak Body Temp (°C) | Fever Onset (Days) | Underlying Disease | Type of HSCT | ES | VOD | Prolonged Fever | GVHD | Conditioning Regimens | Long-Term (>2 Years) Follow-Up Data |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 001-D0 | M | 3Y6M | 38.9 | 6 | β thalassemia/ HbE | MR | No | No | No | No | BUSX+CTX | No long-term effects |
| 001-D3 | 37.4 | 9 | ||||||||||
| 002-DA | M | 12Y1M | - | - | Acute myeloid leukemia | HAP | Yes | No | Yes (day 1–16) | No | FLU+THI+ BUSF | No long-term effects |
| 002-D0 | 39.7 | 1 | ||||||||||
| 002-D3 | 37.6 | 4 | ||||||||||
| 003-D0 | F | 3Y7M | 38.2 | 2 | Neuroblastoma | HAP | Yes | No | No | Yes | BUSF+MEL+ATG | No long-term effects |
| 003-D3 | 37.8 | 5 | ||||||||||
| 004-D0 | M | 3Y2M | 38.5 | 4 | Langerhans cell Histiocytosis | HAP | No | No | Yes (day 1–19) | Yes | FLU+BUSF+ ATG | No long-term effects |
| 004-D3 | 37 | 7 | ||||||||||
| 005-D3 | M | 6Y2M | 37.4 | 11 | Anaplastic large cell lymphoma | AUT | No | No | No | No | BCNU+ETO+CTX | No long-term effects |
| 006-D0 | F | 3Y8M | 40 | 6 | Yolk sac tumor | AUT | No | No | No | No | ETO+CARB+CTX | No long-term effects |
| 006-D3 | 37.1 | 9 | ||||||||||
| 007-D0 | M | 1Y11M | 38.2 | 2 | Acute myeloid leukemia | HAP | No | No | No | Yes | FLU+THI+ BUSF | No long-term effects |
| 007-D3 | 37.4 | 5 | ||||||||||
| 008-D0 | M | 3Y9M | 39.7 | 10 | Chronic granulomatous disease | HAP | No | No | Yes (day 8–18) | Yes | FLU+BUSF+ ATG | Chronic GVHD |
| 008-D3 | 38.4 | 13 | ||||||||||
| 009-D0 | F | 3Y9M | 40 | 3 | Neuroblastoma | HAP | No | No | No | Yes | BUSF+MEL+ATG | No long-term effects |
| 009-D3 | 36.6 | 6 | ||||||||||
| 011-DA | M | 2Y4M | - | - | Wiskott Aldrich Syndrome | MR | No | No | No | No | BUSF+CTX | No long-term effects |
| 011-D0 | 38.1 | 3 | ||||||||||
| 011-D3 | 38.5 | 7 | ||||||||||
| 012-D0 | F | 1Y4M | 38.9 | 2 | Acute lymphocytic leukemia | HAP | Yes | No | No | No | FLU+THI+ BUSF | No long-term effects |
| 012-D3 | 38.0 | 6 | ||||||||||
| 013-D0 | F | 3Y5M | 38.2 | 3 | β thalassemia/ HbE | HAP | Yes | No | No | Yes | FLU+BUSF+ ATG | Death from GVHD and bleeding |
| 013-D3 | 38.3 | 7 | ||||||||||
| 014-DA | F | 3Y3M | - | - | Undifferentiated round cell tumor | AUT | - | - | No | - | BUSX+MEL | Death from RDS on 39 days post-HSCT |
| 015-DA | M | 7Y6M | - | - | Neuroblastoma | MR | No | No | No | No | BUSF+MEL | Neuroblastoma relapse on 164 days post-HSCT |
| 015-D0 | 38.1 | 6 | ||||||||||
| 015-D3 | 37.6 | 9 |
| TE-NGS | Routine Test | Confirmation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample Code | Total Number of Reads (Million) | Viral Reads % | Total Number of Viral Reads | NDV-Internal Control | Identified Viruses | |||||||
| % Genome Coverage | Reads | % a | Virus Identified | % Genome Coverage | Reads | % a | Multiplexed qRT-PCR b | Targeted qRT-PCR b | ||||
| 001-D0 | 1.83 | 21.4 | 391,620 | 100 | 139,882 | 35.7 | - | - | - | - | Neg | NA |
| 001-D3 | 3.76 | 48.4 | 1,819,840 | 100 | 1,737,559 | 95.5 | - | - | - | - | Neg | NA |
| 002-DA | 2.66 | 91.8 | 2,441,880 | 100 | 2,384,116 | 97.6 | HPgV-1 | 94.2 | 33,916 | 1.4 | Neg | HPgV-1 (Ct 28.58) |
| 002-D0 | 2.11 | 28.5 | 601,350 | 100 | 362,876 | 60.3 | HPgV-1 | 98.4 | 31,480 | 0.6 | Neg | HPgV-1 (Ct 29.10) |
| 002-D3 | 4.02 | 59.6 | 2,395,920 | 100 | 2,302,948 | 96.1 | HPgV-1 | 96.5 | 23,319 | 0.9 | Neg | HPgV-1 (Ct 29.54) |
| 003-D0 | 2.46 | 32.0 | 787,200 | 100 | 741,509 | 94.2 | - | - | - | - | Neg | NA |
| 003-D3 | 1.59 | 10.6 | 168,540 | 100 | 5415 | 3.2 | - | - | - | - | Neg | NA |
| 004-D0 | 3.91 | 58.8 | 2,299,080 | 100 | 2,232,001 | 97.1 | - | - | - | - | CMV (Ct 34.07) | NA |
| 004-D3 | 0.73 | 5.5 | 40,150 | 100 | 21,688 | 54.0 | CMV | 67.2 | 2221 | 5.5 | CMV (Ct 31.18) | NA |
| 005-D0 | 1.90 | 16.2 | 307,800 | 100 | 268,477 | 87.2 | - | - | - | - | Neg | NA |
| 006-D0 | 1.71 | 2.3 | 39,330 | 99.5 | 3174 | 8.0 | - | - | - | - | Neg | NA |
| 006-D3 | 2.63 | 42.8 | 1,125,640 | 100 | 1,057,996 | 94.0 | - | - | - | - | Neg | NA |
| 007-D0 | 1.97 | 13.7 | 269,890 | 100 | 69,031 | 25.6 | - | - | - | - | Neg | NA |
| 007-D3 | 1.05 | 82.1 | 862,050 | 100 | 841,928 | 97.7 | - | - | - | - | Neg | NA |
| 008-D0 | 2.06 | 6.8 | 140,080 | 100 | 76,091 | 54.3 | - | - | - | - | CMV (Ct 35.81) | NA |
| 008-D3 | 2.19 | 2.8 | 61,320 | 99.6 | 7035 | 11.5 | - | - | - | - | CMV (Ct 36.89) | NA |
| 009-D0 | 1.97 | 3.2 | 63,040 | 100 | 12,533 | 19.9 | - | - | - | - | Neg | NA |
| 009-D3 | 1.27 | 2.5 | 31,750 | 99.5 | 2916 | 9.2 | - | - | - | - | Neg | NA |
| 011-DA | 5.36 | 69.8 | 3,741,280 | 100 | 1,740,886 | 46.5 | HPgV-1 | 100 | 69,446 | 1.8 | Neg | HPgV-1 (Ct 25.17) |
| 011-D0 | 1.86 | 36.8 | 684,480 | 100 | 603,461 | 88.2 | HPgV-1 | 98.9 | 44,268 | 6.4 | Neg | HPgV-1 (Ct 30.22) |
| 011-D3 | 2.20 | 38.9 | 855,800 | 100 | 758,192 | 88.6 | HPgV-1 | 99.0 | 62,142 | 7.2 | Neg | HPgV-1 (Ct 30.65) |
| 012-D0 | 4.61 | 72.1 | 3,323,810 | 100 | 3,165,402 | 95.2 | - | - | - | - | Neg | NA |
| 013-D0 | 1.49 | 12.7 | 189,230 | 100 | 138,532 | 73.2 | - | - | - | - | Neg | NA |
| 013-D3 | 1.70 | 19.4 | 329,800 | 100 | 102,209 | 31.0 | - | - | - | - | Neg | NA |
| 014-DA | TE-NGS was not performed | Neg | Neg for HPgV-1 | |||||||||
| 015-DA | 1.04 | 15.5 | 161,200 | 100 | 85,325 | 52.9 | - | - | - | - | Neg | Neg for HPgV-1 |
| 015-D0 | 3.40 | 59.4 | 2,019,600 | 100 | 830,550 | 41.1 | HPgV-1 | 97.9 | 58,016 | 2.8 | Neg | HPgV-1 (Ct 26.94) |
| 015-D3 | 0.19 | 43.9 | 83,410 | 100 | 30,992 | 37.1 | HPgV-1 | 98.6 | 3935 | 4.7 | Neg | HPgV-1 (Ct 26.28) |
| Sample Name | Genome Size | Number of Reads | Average Assembly Depth | Accession Number |
|---|---|---|---|---|
| 002-DA | 8714 | 66,577 | 1436 | MZ099565 |
| 002-D0 | 9082 | 54,861 | 1273 | MZ099566 |
| 002-D3 | 9080 | 40,980 | 914 | MZ099567 |
| 011-DA | 9273 | 118,572 | 2764 | MZ099568 |
| 011-D0 | 9112 | 77,867 | 1722 | MZ099569 |
| 011-D3 | 9096 | 106,570 | 2485 | MZ099570 |
| 015-D0 | 9232 | 98,765 | 2268 | MZ099571 |
| 015-D3 | 9336 | 6691 | 154 | MZ099572 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludowyke, N.; Phumiphanjarphak, W.; Apiwattanakul, N.; Manopwisedjaroen, S.; Pakakasama, S.; Sensorn, I.; Pasomsub, E.; Chantratita, W.; Hongeng, S.; Aiewsakun, P.; et al. Target Enrichment Metagenomics Reveals Human Pegivirus-1 in Pediatric Hematopoietic Stem Cell Transplantation Recipients. Viruses 2022, 14, 796. https://doi.org/10.3390/v14040796
Ludowyke N, Phumiphanjarphak W, Apiwattanakul N, Manopwisedjaroen S, Pakakasama S, Sensorn I, Pasomsub E, Chantratita W, Hongeng S, Aiewsakun P, et al. Target Enrichment Metagenomics Reveals Human Pegivirus-1 in Pediatric Hematopoietic Stem Cell Transplantation Recipients. Viruses. 2022; 14(4):796. https://doi.org/10.3390/v14040796
Chicago/Turabian StyleLudowyke, Natali, Worakorn Phumiphanjarphak, Nopporn Apiwattanakul, Suwimon Manopwisedjaroen, Samart Pakakasama, Insee Sensorn, Ekawat Pasomsub, Wasun Chantratita, Suradej Hongeng, Pakorn Aiewsakun, and et al. 2022. "Target Enrichment Metagenomics Reveals Human Pegivirus-1 in Pediatric Hematopoietic Stem Cell Transplantation Recipients" Viruses 14, no. 4: 796. https://doi.org/10.3390/v14040796
APA StyleLudowyke, N., Phumiphanjarphak, W., Apiwattanakul, N., Manopwisedjaroen, S., Pakakasama, S., Sensorn, I., Pasomsub, E., Chantratita, W., Hongeng, S., Aiewsakun, P., & Thitithanyanont, A. (2022). Target Enrichment Metagenomics Reveals Human Pegivirus-1 in Pediatric Hematopoietic Stem Cell Transplantation Recipients. Viruses, 14(4), 796. https://doi.org/10.3390/v14040796