
An Evaluation of Phylogenetic Workflows in Viral Molecular Epidemiology



Abstract
:1. Introduction
2. Methods
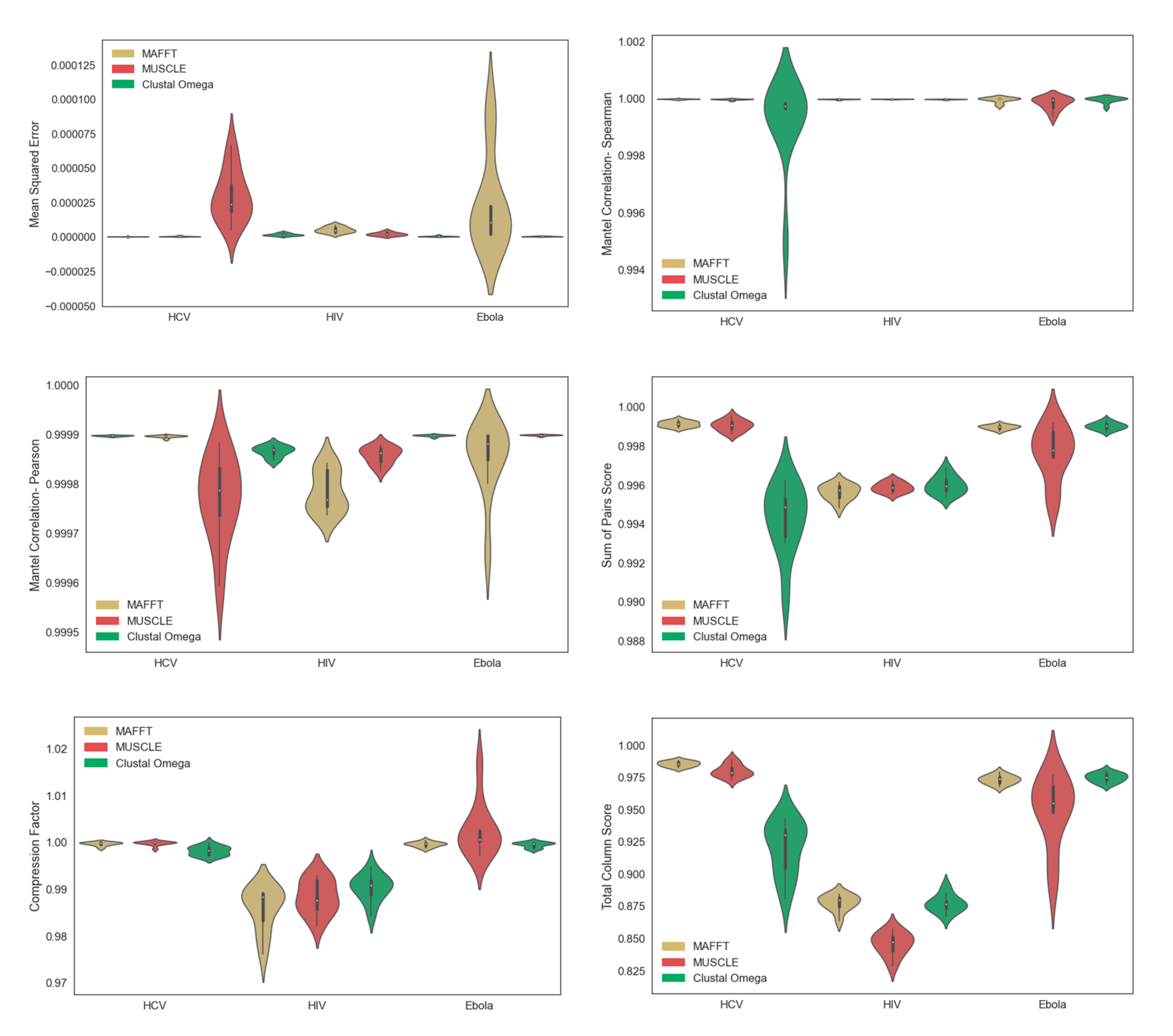
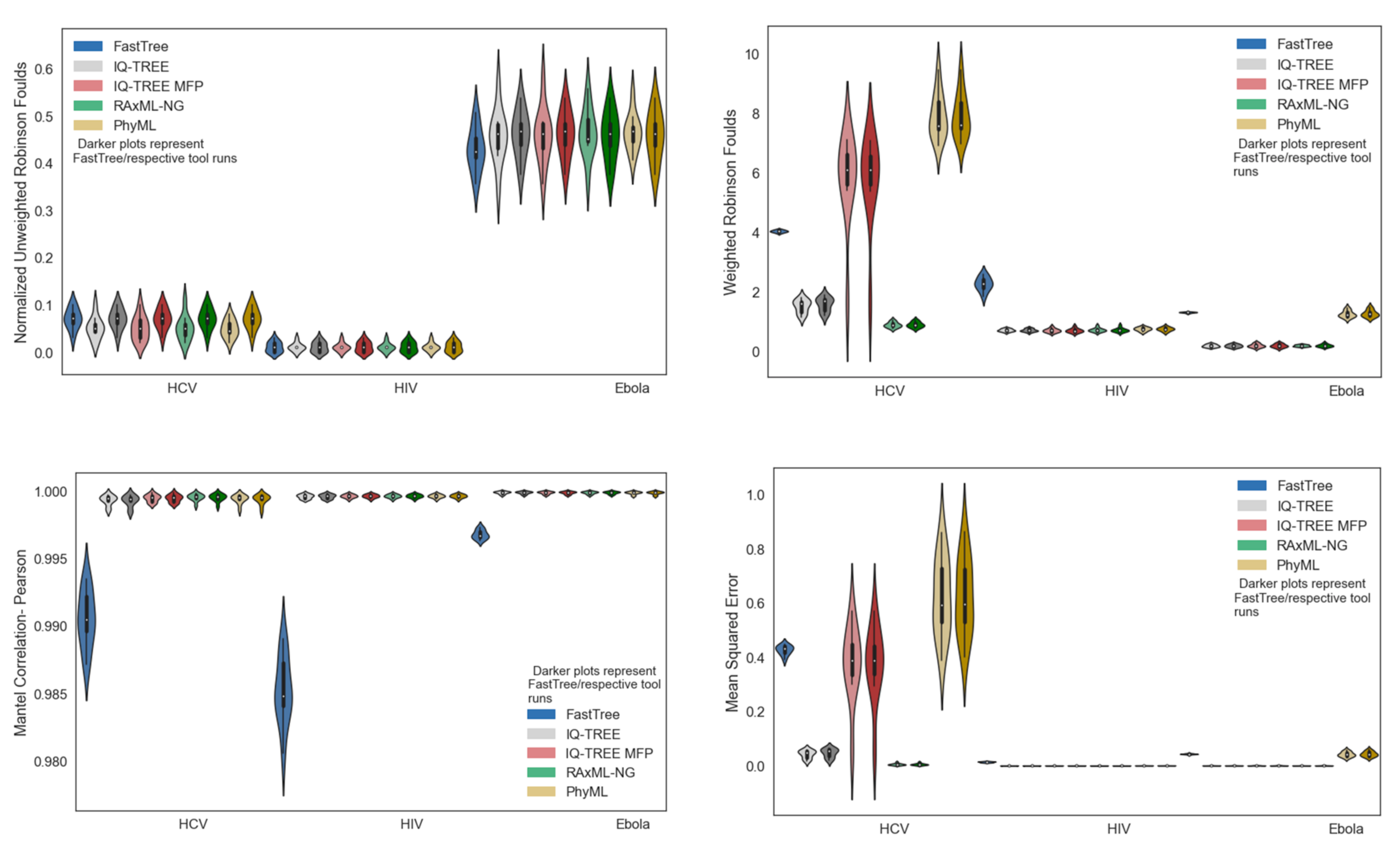
2.1. Mean Squared Error
2.2. Mantel Correlation
2.3. Robinson–Foulds (RF) Distance
2.4. Sum of Pairs (SP) Score
2.5. Total Columns (TC) Score
2.6. Compression Factor
3. Results
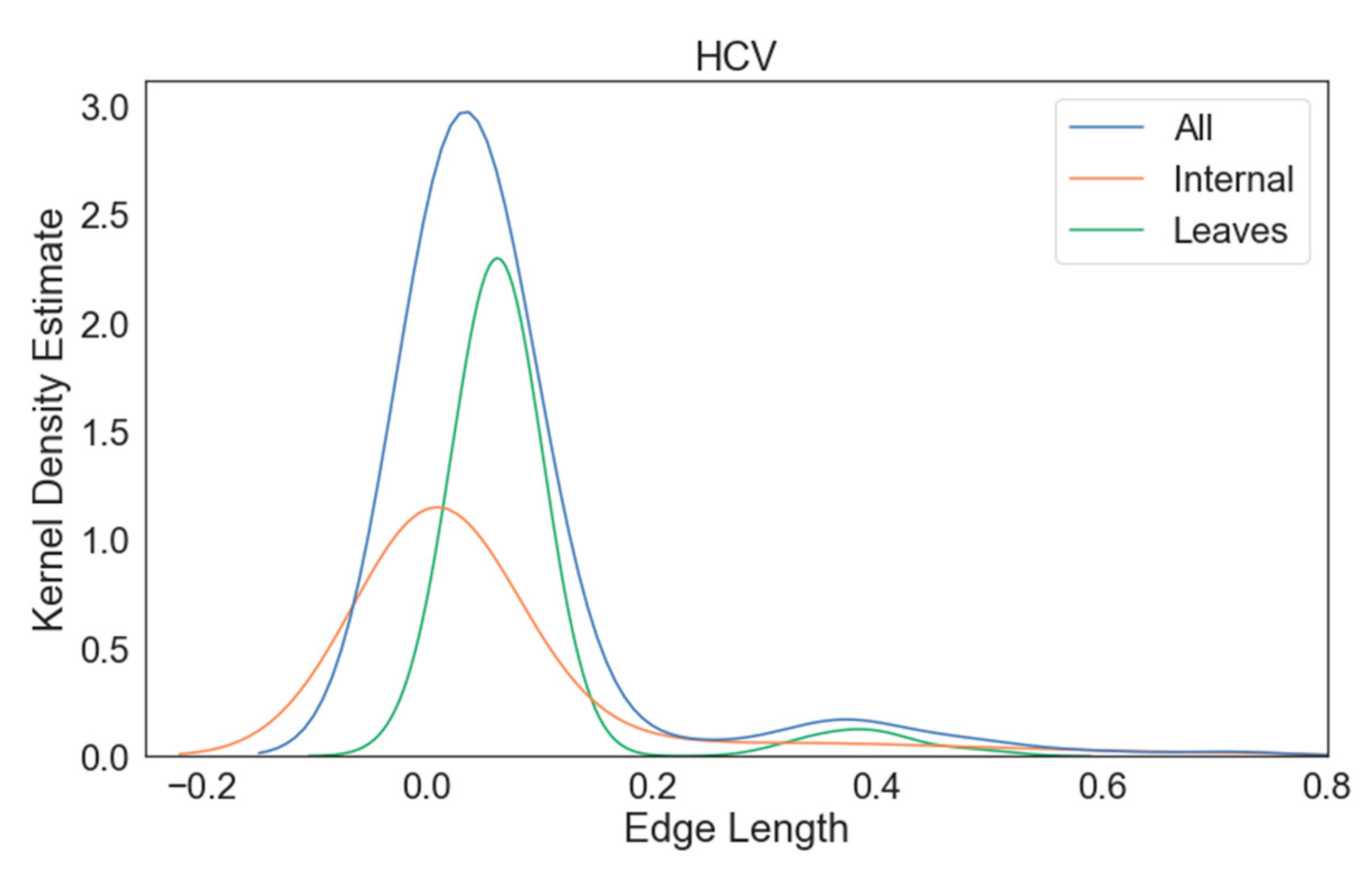
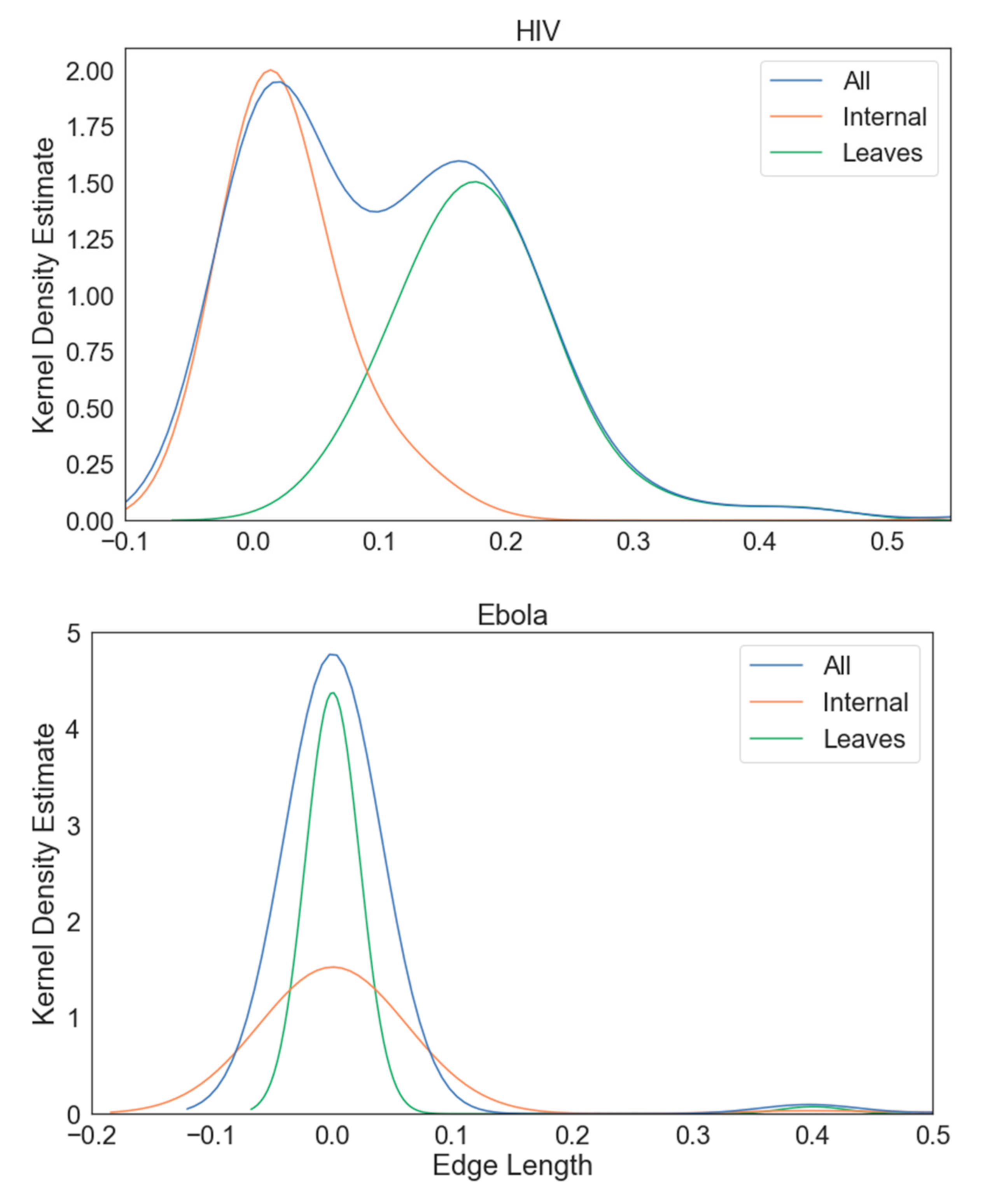
3.1. Multiple Sequence Alignment
3.2. Phylogenetic Inference
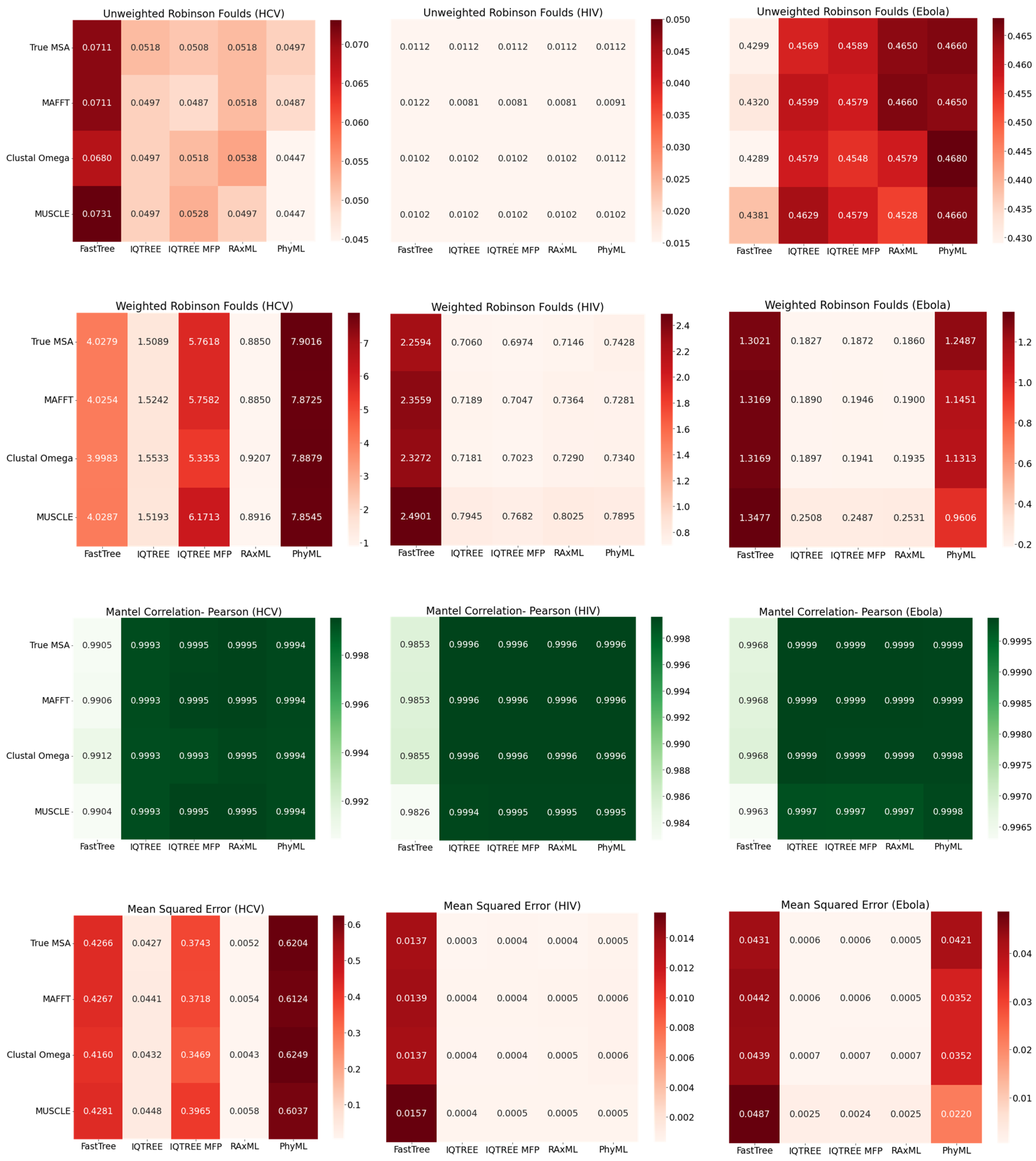
3.3. Combinations of MSA and Phylogenetic Inference
3.4. Combinations of MSA and Optimized FastTree Topologies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, M.; Moshiri, N.; Mai, U.; Jia, X.; Mirarab, S. TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS ONE 2019, 14, e0221068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.; Lycett, S. UK HIV Drug Resistance Database. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosperi, M.C.; Ciccozzi, M.; Fanti, I.; Saladini, F.; Pecorari, M.; Borghi, V.; Di Giambenedetto, S.; Bruzzone, B.; Capetti, A.; Vivarelli, A.; et al. A novel methodology for large-scale phylogeny partition. Nat. Commun. 2011, 2, 321. [Google Scholar] [CrossRef] [Green Version]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2016, 17, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Chernomor, O.; Von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [PubMed] [Green Version]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Mai, U.; Sayyari, E.; Mirarab, S. Minimum variance rooting of phylogenetic trees and implications for species tree reconstruction. PLoS ONE 2017, 12, e0182238. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, W.; Yang, Z. INDELible: A Flexible Simulator of Biological Sequence Evolution. Mol. Biol. Evol. 2009, 26, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. A space-time process model for the evolution of DNA sequences. Genetics 1995, 139, 993–1005. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Shen, X.X.; Hittinger, C.T.; Rokas, A. Evaluating Fast Maximum Likelihood-Based Phylogenetic Programs Using Empirical Phylogenomic Data Sets. Mol. Biol. Evol. 2018, 35, 486–503. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar] [PubMed]
- Robinson, D.F.; Foulds, L.R. Comparison of phylogenetic trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Mirarab, S.; Warnow, T. FastSP: Linear time calculation of alignment accuracy. Bioinformatics 2011, 27, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Linder, C.R.; Warnow, T. RAxML and FastTree: Comparing Two Methods for Large-Scale Maximum Likelihood Phylogeny Estimation. PLoS ONE 2011, 6, e27731. [Google Scholar] [CrossRef]
- Martyn, I.; Steel, M. The impact and interplay of long and short branches on phylogenetic information content. J. Theor. Biol. 2012, 314, 157–163. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, A.; Sereda, P.; Brumme, C.J.; Brumme, Z.L.; Barrios, R.; Montaner, J.S.G.; Joy, J.B. Concordance of HIV transmission risk factors elucidated using viral diversification rate and phylogenetic clustering. Evol. Med. Public Health 2021, 9, 338–348. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Seconds) | FastTree 2 | RaxML | PhyML | IQ-TREE | IQ-TREE MFP |
|---|---|---|---|---|---|
| Total | 645 | >604,800 | memory | 84,931 | 266,399 (142,286 MFP) |
| BL optimization | N/A | 757 | memory | 1532 | 4885 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, C.; Meng, S.; Moshiri, N. An Evaluation of Phylogenetic Workflows in Viral Molecular Epidemiology. Viruses 2022, 14, 774. https://doi.org/10.3390/v14040774
Young C, Meng S, Moshiri N. An Evaluation of Phylogenetic Workflows in Viral Molecular Epidemiology. Viruses. 2022; 14(4):774. https://doi.org/10.3390/v14040774
Chicago/Turabian StyleYoung, Colin, Sarah Meng, and Niema Moshiri. 2022. "An Evaluation of Phylogenetic Workflows in Viral Molecular Epidemiology" Viruses 14, no. 4: 774. https://doi.org/10.3390/v14040774
APA StyleYoung, C., Meng, S., & Moshiri, N. (2022). An Evaluation of Phylogenetic Workflows in Viral Molecular Epidemiology. Viruses, 14(4), 774. https://doi.org/10.3390/v14040774