
Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil

 , , , , , , , , , , , , , ,
, , , , , , , , , , , , , ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
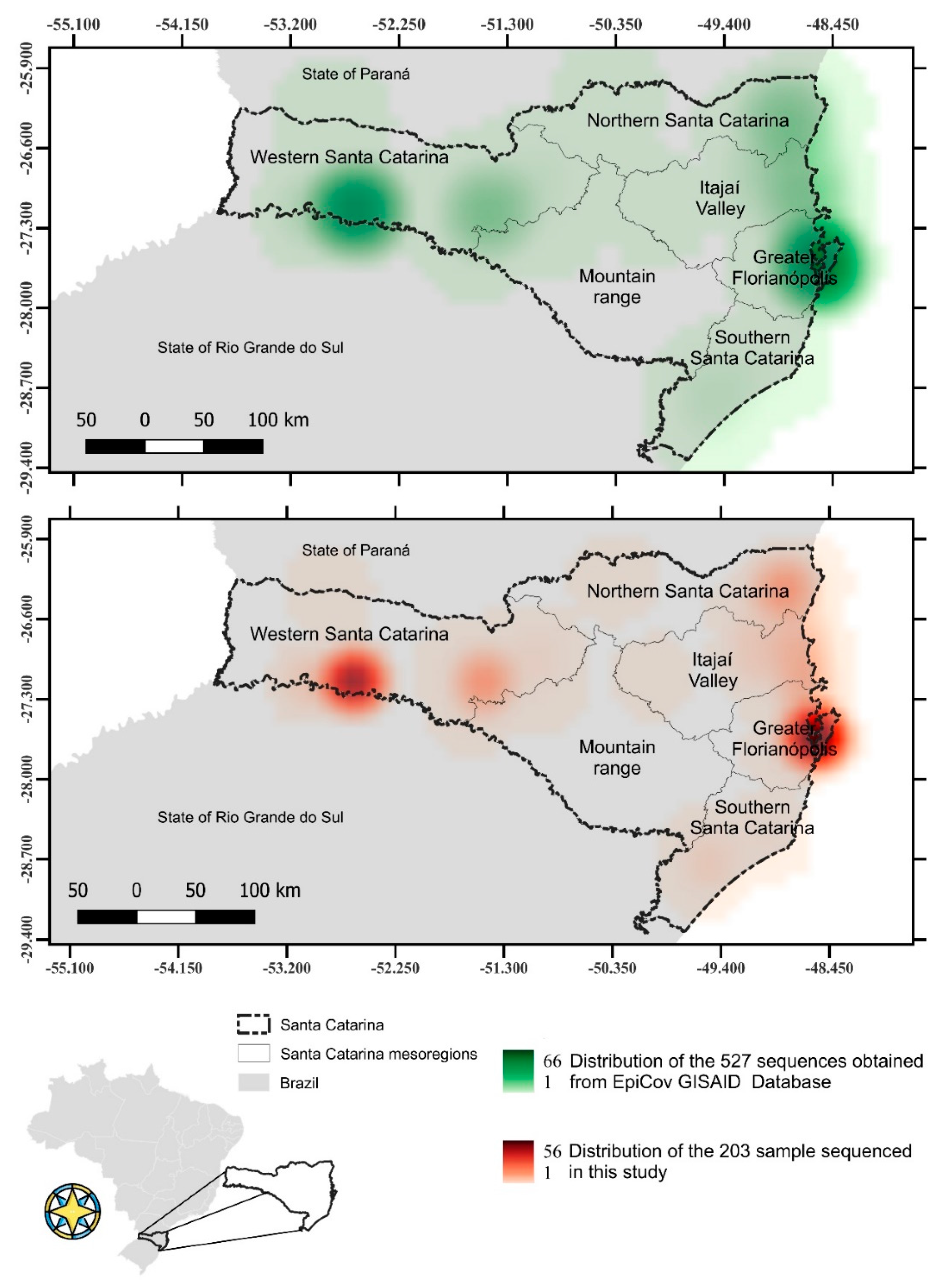
2.1. Studied Regions and Sampling
2.2. RNA Purification and Sequencing
2.3. SARS-CoV-2 Genome Assembling and Variant Analysis
2.4. Maximum-Likelihood (ML) Phylogenetic Analysis
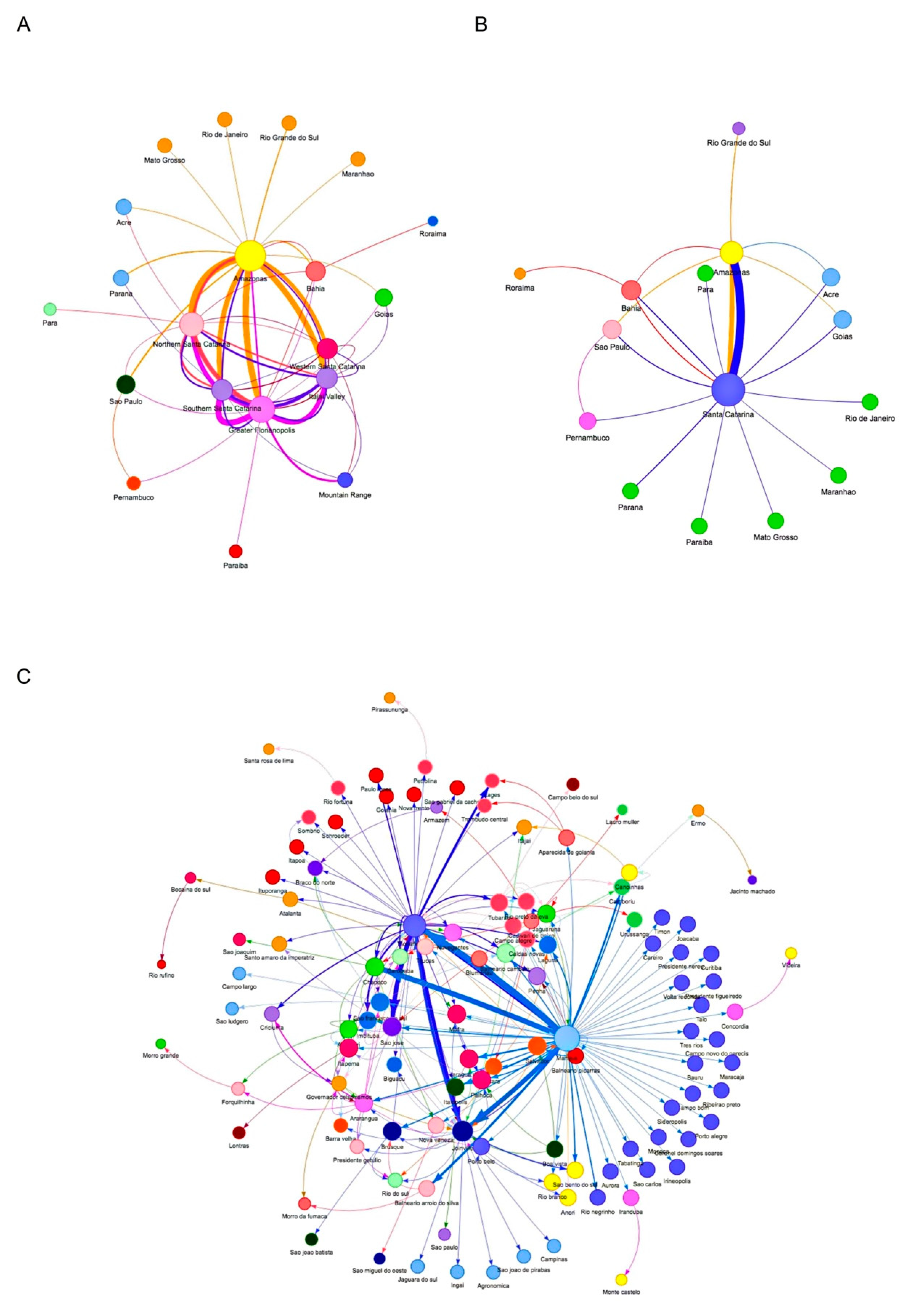
2.5. Network Analysis of SC VOC Gamma and Related Lineages during the Second Wave
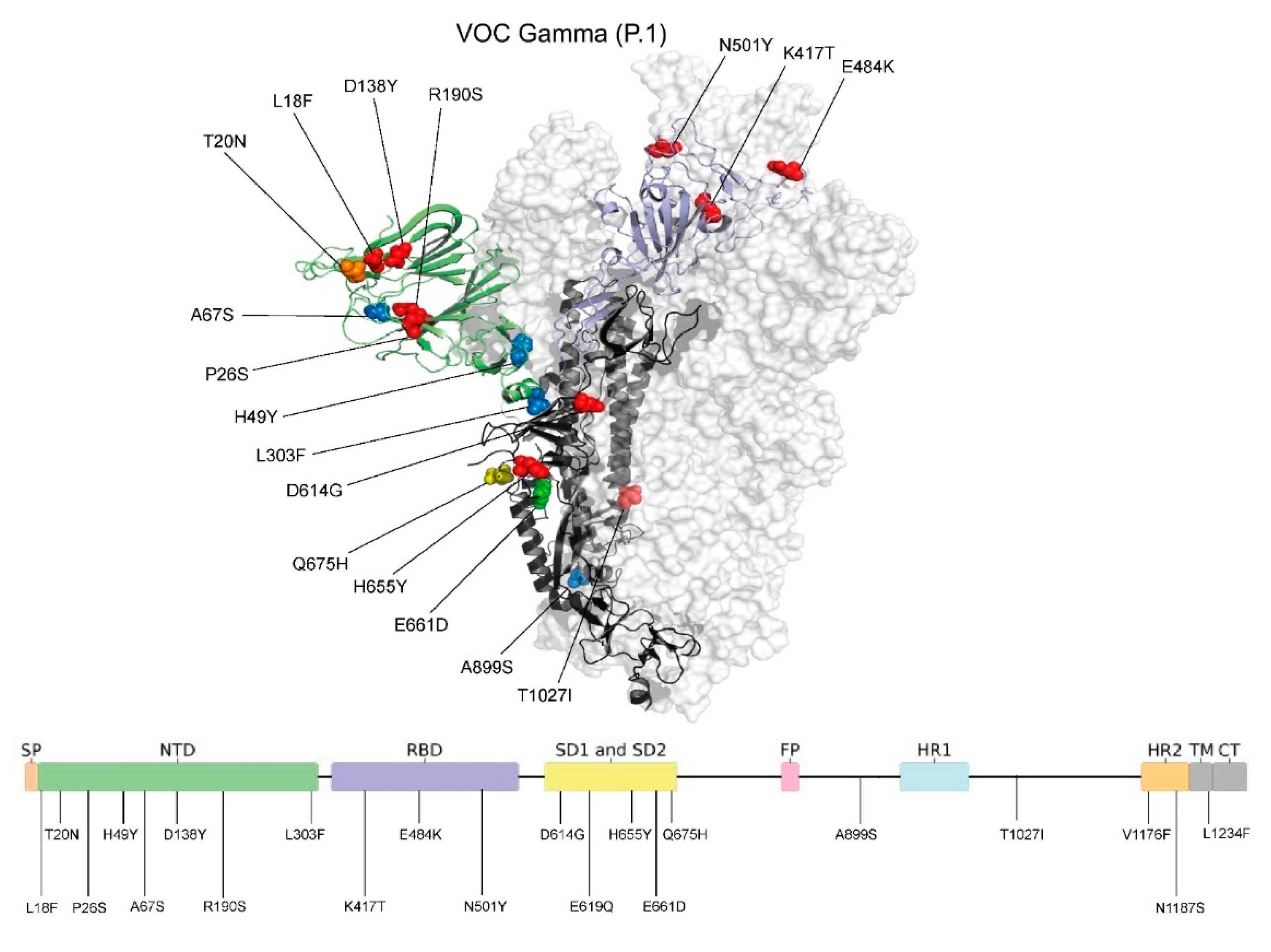
2.6. Spike (S) Gene Sequence Polymorphism and Protein Structure Analysis
3. Results
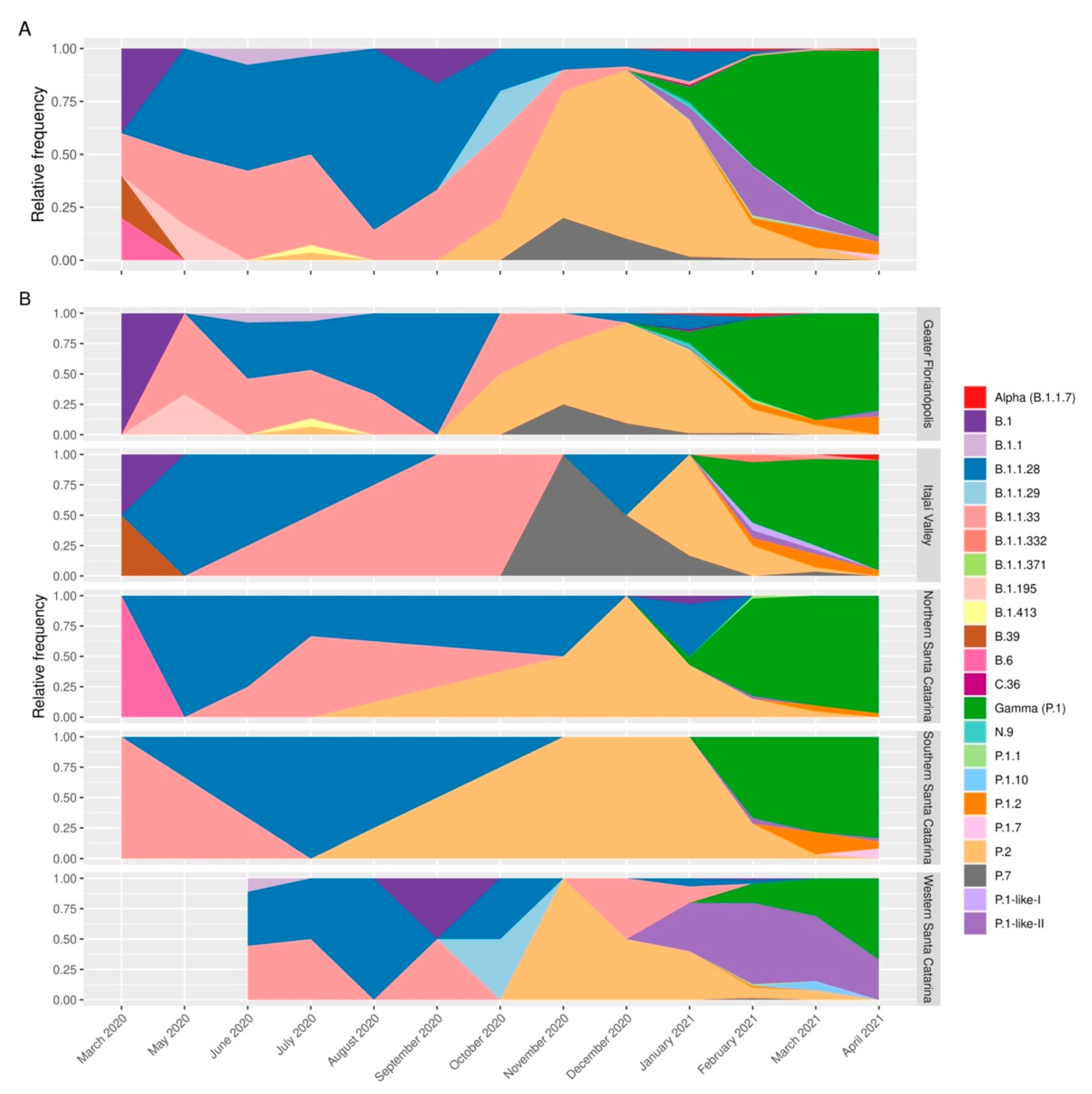
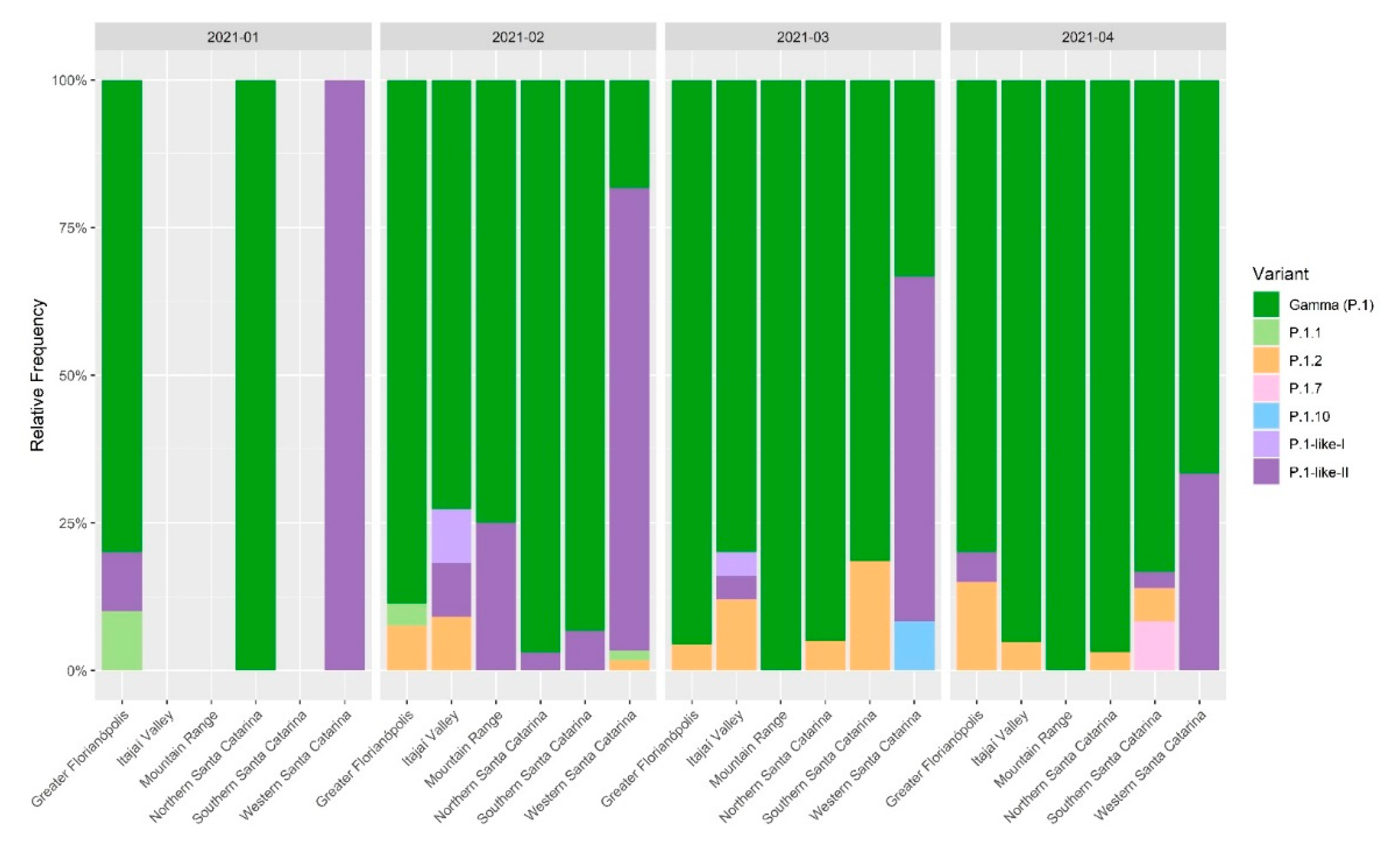
3.1. Profile of SARS-CoV-2 Variants in the State of Santa Catarina after One Year of the COVID-19 Pandemic
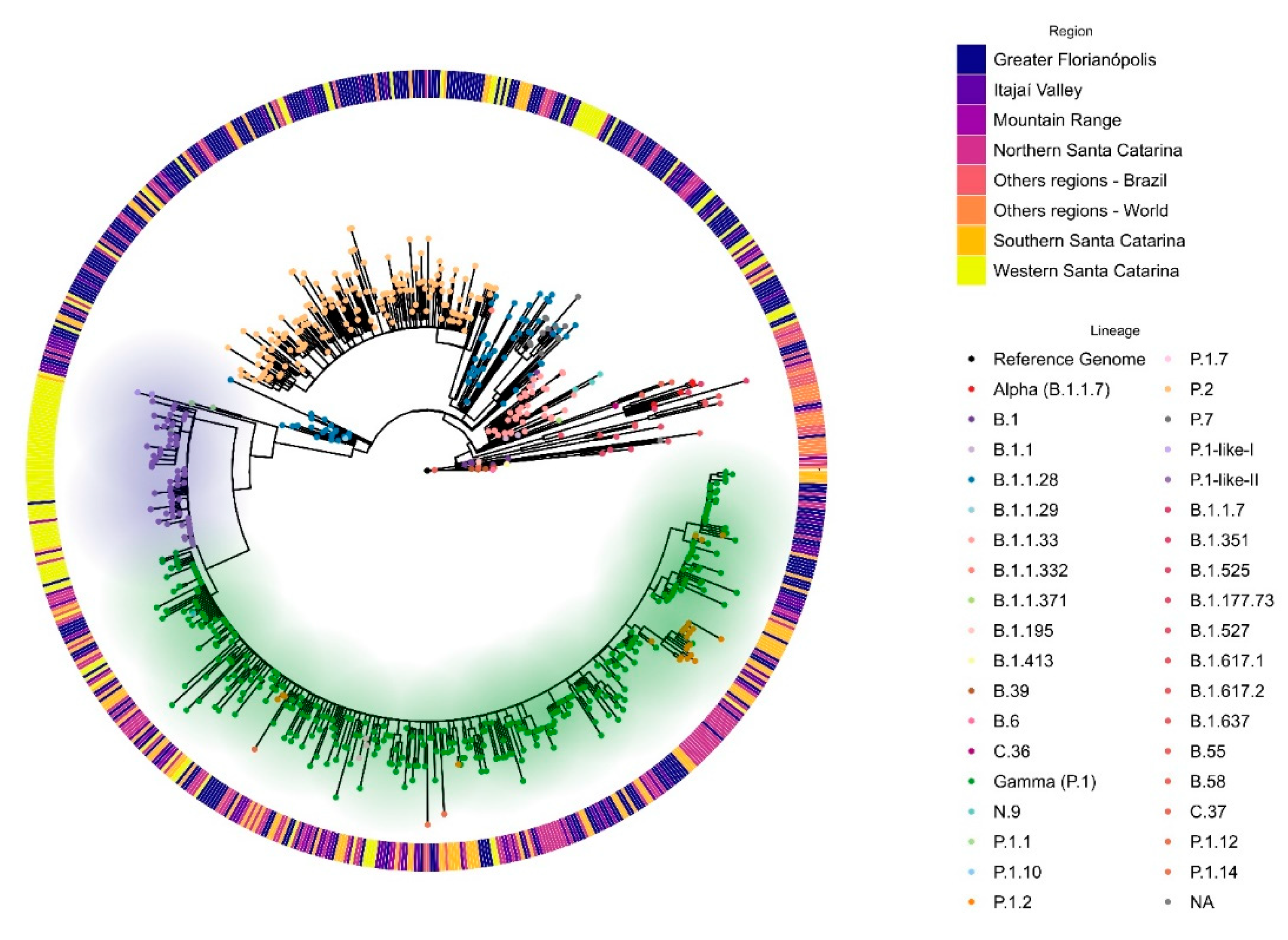
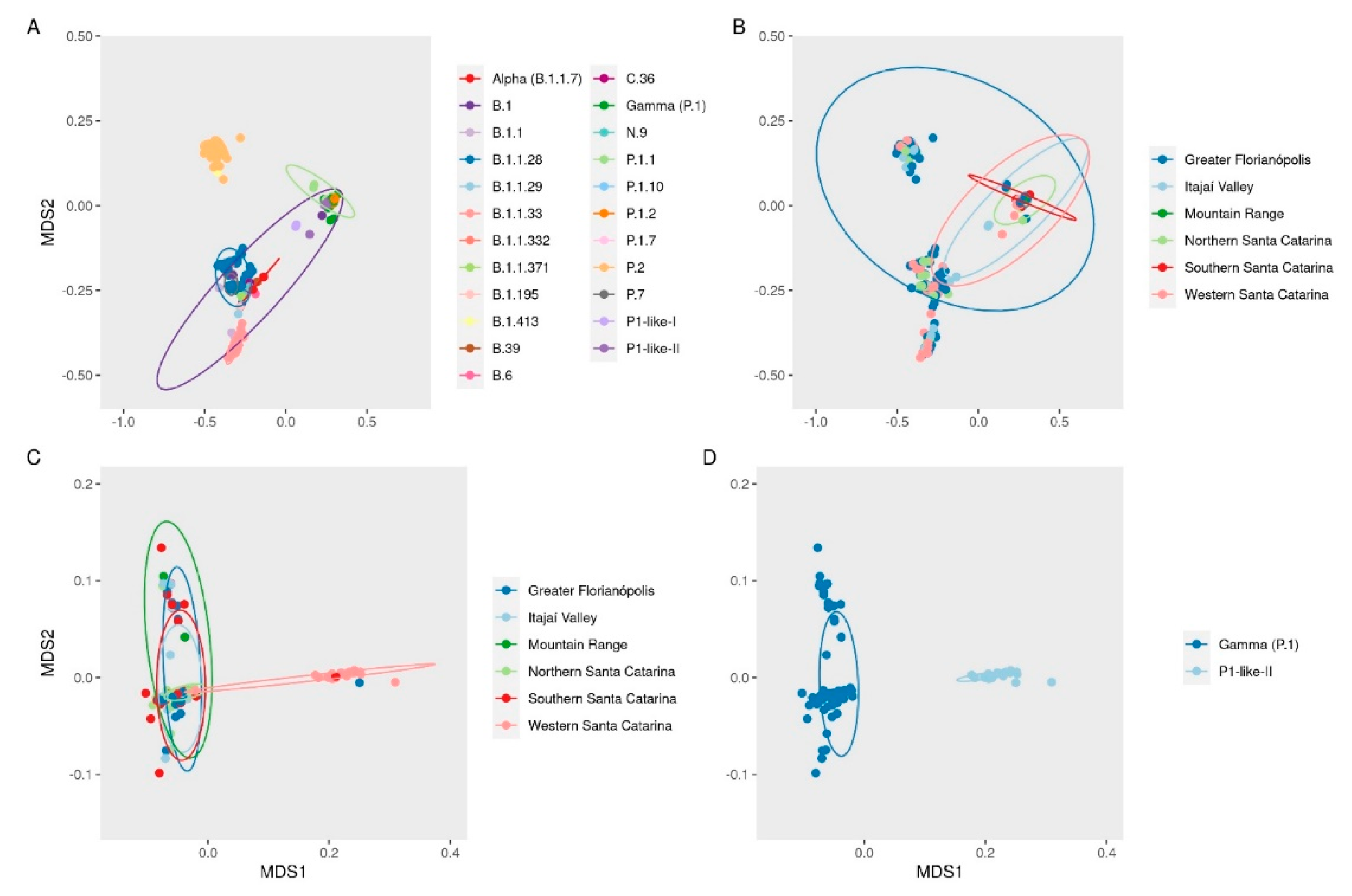
3.2. VOC Gamma and Related Lineages in Santa Catarina
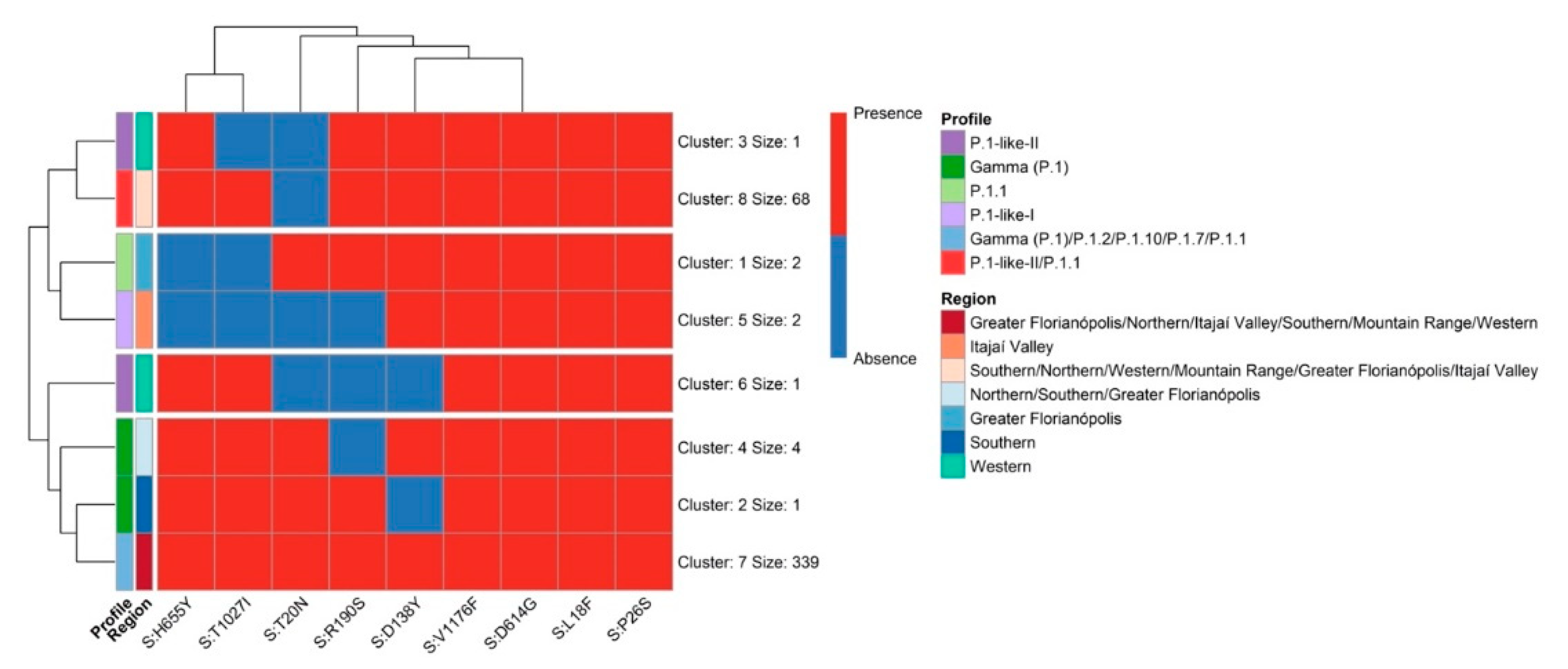
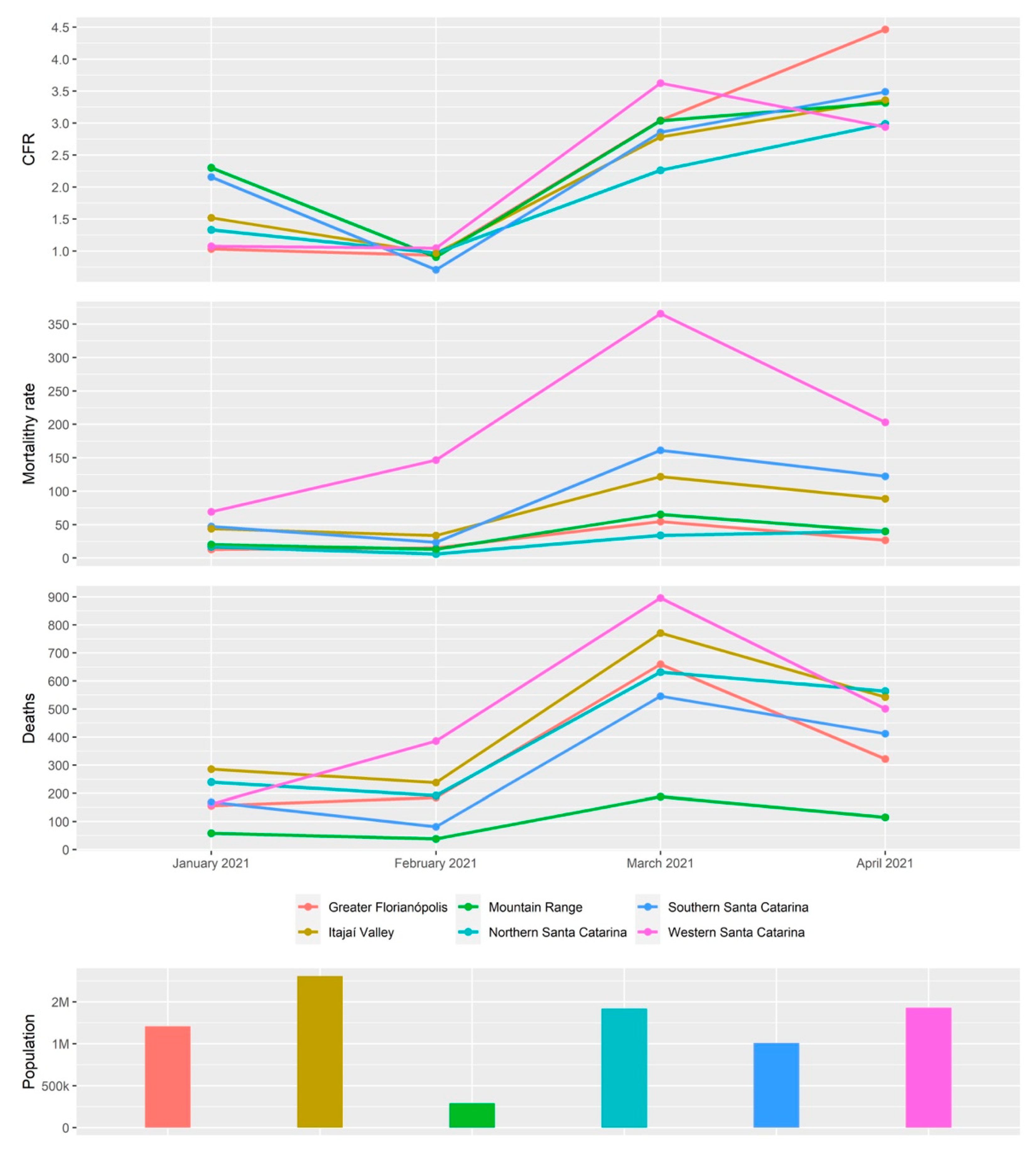
3.3. The Rapid Spread and Regionalization of P.1-like-II during the Second Wave of the COVID-19 Pandemic in Santa Catarina, Brazil
4. Discussion
5. Conclusions
Disclaimer
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus (COVID-19). Available online: https://www.who.int (accessed on 12 December 2021).
- National Council of Health Secretaries-Covid-19 CONASS Panel. Available online: https://www.conass.org.br/painelconasscovid19/ (accessed on 4 December 2021).
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of healthcare associated COVID-19: A prospective genomic surveillance study. Lancet Infect. Dis. 2021, 21, e36, Corrected and republished from Lancet Infect Dis. 2020, 20, 1263–1272. [Google Scholar]
- Global Initiative on Sharing Avian Influenza Data. 2008. Available online: https://www.gisaid.org (accessed on 27 November 2021).
- Tao, K.; Tzou, P.L.; Nouhin, J. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S. Genomics and epidemiology of P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, C.J.; Löbermann, M.; Reisinger, E.C. COVID-19: Epidemiology and mutations: An update [in German]. Radiologe 2021, 61, 880–887. [Google Scholar] [CrossRef]
- Nonaka, C.K.V.; Gräf, T.; Barcia, C.A.L.; Costa, V.F.; de Oliveira, J.L.; Passos, R.D.H. SARS-CoV-2 variant of concern P.1 (Gamma) infection in young and middle-aged patients admitted to the intensive care units of a single hospital in Salvador, Northeast Brazil, February 2021. Int. J. Infect. Dis. 2021, 111, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe. 2021, 29, 44–57. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Gräf, T.; Bello, G.; Venas, T.M.M.; Pereira, E.C.; Paixão, A.C.D.; Appolinario, L.R. Identification of SARS-CoV-2 P.1-Related Lineages in Brazil Provides New Insights about the Mechanisms of Emergence of Variants of Concern. 2021. Available online: https://doi.org/10.21203/rs.3.rs-580195/v1 (accessed on 30 November 2021).
- Varela, A.P.M.; Prichula, J.; Mayer, F.Q.; Salvato, R.S.; Sant’Anna, F.H.; Gregianini, T.S.; Martins, L.G.; Seixas, A.; Veiga, A.B.G.D. SARS-CoV-2 introduction and lineage dynamics across three epidemic peaks in Southern Brazil: Massive spread of P.1. Infect. Genet. Evol. 2021, 96, 105144. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Schemberger, M.O.; Suzukawa, A.A.; Riediger, I.N.; do Carmo Debur, M.; Becker, G. Re-emergence of Gamma-like-II and emergence of Gamma-S:E661D SARS-CoV-2 lineages in the south of Brazil after the 2021 outbreak. Virol. J. 2021, 18, 222. [Google Scholar] [CrossRef]
- Santa Catarina Epidemiological Surveillance Directorate. 2020. Available online: https://www.dive.sc.gov.br/ (accessed on 7 November 2021).
- Brazilian Institute of Geography and Statistics-Official Territorial Area-Federative Unit Consultation. 2010. Available online: https://www.ibge.gov.br/cidades-e-estados/sc.html (accessed on 15 October 2021).
- Eden, J.-S.; Sim, E. SARS-CoV-2 Genome Sequencing Using Long Pooled Amplicons on Illumina Platforms. 2020. Available online: https://www.protocols.io/view/sars-cov-2-genome-sequencing-using-long-pooled-amp-befyjbpw?step=81 (accessed on 24 November 2021).
- Young, E.; Oakeson, K. Utah DoH ARTIC/Illumina Bioinformatic Workflow. 2021. Available online: https://github.com/CDCgov/SARS-CoV-2_Sequencing/tree/master/protocols/BFX-UT_ARTIC_Illumina (accessed on 28 November 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Nextclade Web 1.10.0. 2020. Available online: https://clades.nextstrain.org/ (accessed on 9 November 2021).
- Pangolin COVID-19 Lineage Assigner. 2021. Available online: https://pangolin.cog-uk.io/ (accessed on 9 November 2021).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D. Vegan: Community Ecology Package. R Package Version 2.0-2. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 5 December 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis Cheatsheet; Springer: New York, NY, USA, 2016. [Google Scholar]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.B.; Ford, C.T.; Hostager, R.; Williams, J.; Cioce, M.; Çatalyürek, Ü.V. StrainHub: A phylogenetic tool to construct pathogen transmission networks. Bioinformatics 2020, 36, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.0. 2017. Available online: https://pymol.org/2/ (accessed on 17 November 2021).
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G. Phylogenetic Relationship of SARS-CoV-2 Sequences from Amazonas with Emerging Brazilian Variants Harboring Mutations E484K and N501Y in the Spike Protein. Genom. Epidemiol. 2021. Available online: https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585 (accessed on 12 November 2021).
- Castro, M.C. Spatiotemporal pattern of COVID-19 spread in Brazil. Science 2021, 372, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.L.; Nascimento, F.; Silva, G. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Lamarca, A.P.; de Almeida, L.G.P.; Francisco, R.D.S., Jr.; Lima, L.F.A.; Scortecci, K.C.; Perez, V.P. Genomic surveillance of SARS-CoV-2 tracks early interstate transmission of P.1 lineage and diversification within P.2 clade in Brazil. PLoS Negl. Trop. Dis. 2021, 15, e0009835. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [Green Version]
- Dejnirattisai, W. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef]
- Giovanetti, M.; Benedetti, F.; Campisi, G.; Ciccozzi, A.; Fabris, S.; Ceccarelli, G.; Tambone, V.; Caruso, A.; Angeletti, S.; Zella, D.; et al. Evolution patterns of SARS-CoV-2: Snapshot on its genome variants. Biochem. Biophys. Res. Commun. 2021, 538, 88–91. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambau, A.T.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Li, D.; Zhang, N.; Bai, G.; Yuan, K.; Xiao, H. O-glycosylation pattern of the SARS-CoV-2 spike protein reveals an “O-Follow-N” rule. Cell Res. 2021, 31, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Arnab, S.; Alok, K.C.; Shanta, D. Covid-19. Infection in India: A Comparative Analysis of the Second Wave with the First Wave. Pathogens 2021, 10, 1222. [Google Scholar]
- Katarzyna, J.; Samuel, A.; Pascal, A.; Mondher, T. On the association between SARS-COV-2 variants and COVID-19 mortality during the second wave of the pandemic in Europe. J. Mark. Access Health Policy 2021, 9, 2002008. [Google Scholar]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A., Jr.; Crispim, M.A.E.; Fraiji, N.A. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Flaxman, S.; Mishra, S.; Gandy, A.; Unwin, H.J.T.; Mellan, T.A.; Coupland, H. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 2020, 584, 257–261. [Google Scholar] [CrossRef]
- da Silva, S.J.R.; Pena, L. Collapse of the public health system and the emergence of new variants during the second wave of the COVID-19 pandemic in Brazil. One Health 2021, 13, 100287. [Google Scholar] [CrossRef]
- Kupek, E. Low COVID-19 vaccination coverage and high COVID-19 mortality rates in Brazilian elderly. Rev. Bras. Epidemiol. 2021, 24, e210041. [Google Scholar] [CrossRef]
- Lumley, S.F.; O’Donnell, D.; Stoesser, N.E.; Matthews, P.C.; Howarth, A.; Hatch, S.B. Antibody Status and Incidence of SARS-CoV-2 Infection in Health Care Workers. N. Engl. J. Med. 2021, 384, 533–540. [Google Scholar] [CrossRef]
- Malta, M.; Strathdee, S.A.; Garcia, P.J. The Brazilian tragedy: Where patients living at the ‘Earth’s lungs’ die of asphyxia, and the fallacy of herd immunity is killing people. EClinicalMedicine 2021, 32, 100757. [Google Scholar] [CrossRef] [PubMed]
- Furlan, L.; Caramelli, B. The regrettable story of the “Covid Kit” and the “Early Treatment of Covid-19” in Brazil. Lancet Reg. Health Am. 2021, 4, 100089. [Google Scholar] [CrossRef] [PubMed]
- Abella, B.S.; Jolkovsky, E.L.; Biney, B.T.; Uspal, J.E.; Hyman, M.C.; Frank, I. Efficacy and Safety of Hydroxychloroquine vs. Placebo for Pre-exposure SARS-CoV-2 Prophylaxis Among Health Care Workers-A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.; Stegemann, M.; Metzendorf, M.I.; Gould, S.; Kranke, P.; Meybohm, P.; Skoetz, N.; Weibel, S. Ivermectin for preventing and treating COVID-19. Cochrane Database Syst. Rev. 2021, 7, CD015017. [Google Scholar]
- Garcia, L.P.; Duarte, E. Nonpharmaceutical interventions for tackling the COVID-19 epidemic in Brazil. Epidemiol. Serv. Saude. 2020, 29, e2020222. [Google Scholar]
- Djin-Ye, O.; Silke, B.; Barbara, B.; Janine, R.; Frank, S.; Susanne, D.; Marianne, W.; von Kleist, M.; Mielke, M.; Thorsten, W.; et al. Trends in respiratory virus circulation following COVID-19-targeted nonpharmaceutical interventions in Germany, January–September 2020: Analysis of national surveillance data. Lancet Reg. Health Eur. 2021, 6, 100112. [Google Scholar]
- Fongaro, G.; Stoco, P.H.; Souza, D.S.M.; Grisard, E.C.; Magri, M.E.; Rogovski, P.; Schörner, M.A.; Barazzetti, F.H.; Christoff, A.P.; de Oliveira, L.F.V.; et al. The presence of SARS-CoV-2 RNA in human sewage in Santa Catarina, Brazil, November 2019. Sci. Total Environ. 2021, 778, 146198. [Google Scholar] [CrossRef]
- Wang, H.; Miller, J.A.; Vergherse, M.; Sibai, M.; Solis, D.; Mfuh, K.O. Multiplex SARS-CoV-2 Genotyping Reverse Transcriptase PCR for Population-Level Variant Screening and Epidemiologic Surveillance. J. Clin. Microbiol. 2021, 59, e0085921. [Google Scholar] [CrossRef]
- Wang, H.; Jean, S.; Eltringham, R.; Madison, J.; Snyder, P.; Tu, H. Mutation-specific SARS-CoV-2 PCR screen: Rapid and accurate detection of variants of concern and the identification of a newly emerging variant with spike L452R mutation. J. Clin. Microbiol. 2021, 59, e0092621. [Google Scholar] [CrossRef]
- Yu, C.Y.; Chan, K.G.; Yean, C.Y.; Ang, G.Y. Nucleic Acid-Based Diagnostic Tests for the Detection SARS-CoV-2: An Update. Diagnostics 2021, 11, 53. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padilha, D.A.; Benetti Filho, V.; Moreira, R.S.; Soratto, T.A.T.; Maia, G.A.; Christoff, A.P.; Barazzetti, F.H.; Schörner, M.A.; Ferrari, F.L.; Martins, C.L.; et al. Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil. Viruses 2022, 14, 695. https://doi.org/10.3390/v14040695
Padilha DA, Benetti Filho V, Moreira RS, Soratto TAT, Maia GA, Christoff AP, Barazzetti FH, Schörner MA, Ferrari FL, Martins CL, et al. Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil. Viruses. 2022; 14(4):695. https://doi.org/10.3390/v14040695
Chicago/Turabian StylePadilha, Dayane Azevedo, Vilmar Benetti Filho, Renato Simões Moreira, Tatiany Aparecida Teixeira Soratto, Guilherme Augusto Maia, Ana Paula Christoff, Fernando Hartmann Barazzetti, Marcos André Schörner, Fernanda Luiza Ferrari, Carolina Leite Martins, and et al. 2022. "Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil" Viruses 14, no. 4: 695. https://doi.org/10.3390/v14040695
APA StylePadilha, D. A., Benetti Filho, V., Moreira, R. S., Soratto, T. A. T., Maia, G. A., Christoff, A. P., Barazzetti, F. H., Schörner, M. A., Ferrari, F. L., Martins, C. L., Kawagoe, E. K., Wachter, J. K., Sachet, P., Baptistella, A. R., Schlindwein, A. D., Coelho, B. K., Fernandes, S. B., Rovaris, D. B., Debiasi dos Anjos, M. P., ... Wagner, G. (2022). Emergence of Two Distinct SARS-CoV-2 Gamma Variants and the Rapid Spread of P.1-like-II SARS-CoV-2 during the Second Wave of COVID-19 in Santa Catarina, Southern Brazil. Viruses, 14(4), 695. https://doi.org/10.3390/v14040695