
New Insights into the Structure and Assembly of Bacteriophage P1


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Phages
2.2. Production of Phage Lysates
2.3. Transmission Electron Microscopy
2.4. pBAD24g Plasmid Construction
2.5. Generation of Single-Gene Knockout Mutants
2.6. Generation of DarB-tetR-mCherry Capsid Localization Sequence Fusion Protein
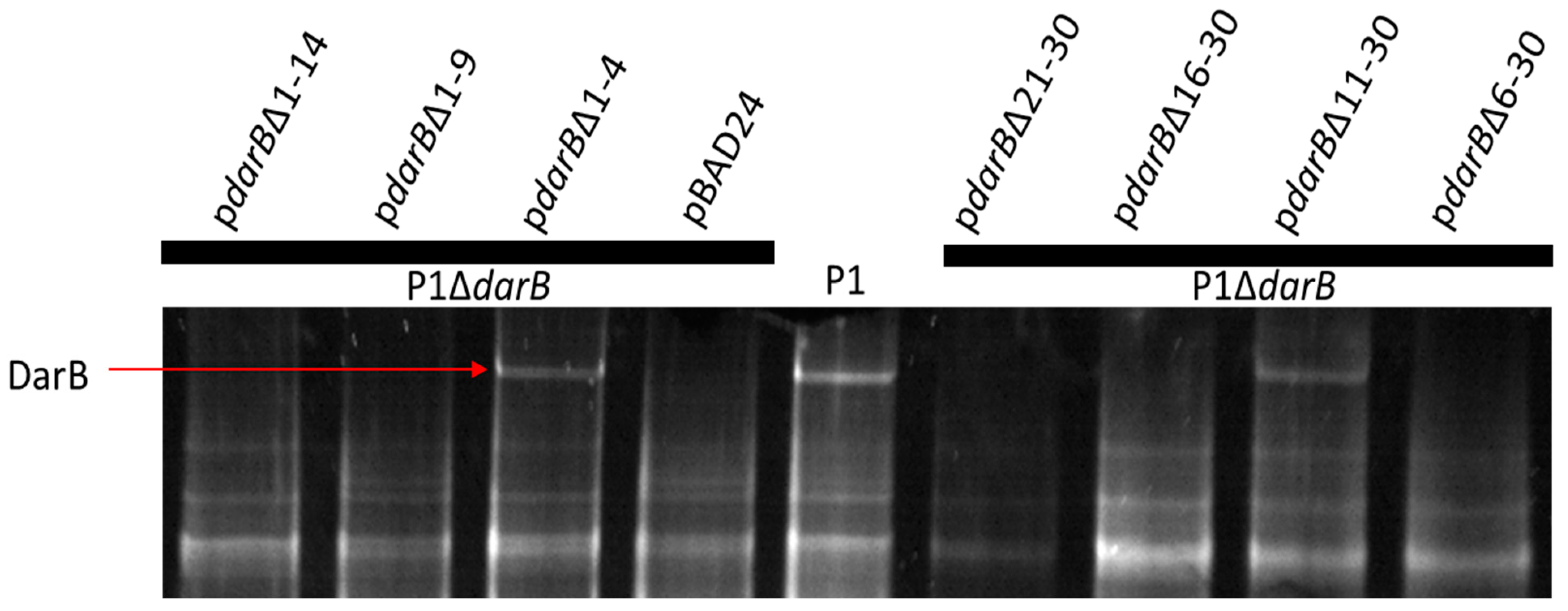
2.7. Generation of N-terminal DarB Mutants
2.8. Generation of Internal DarB Truncation Mutants
2.9. Efficiency of Plating (EOP) Assays
2.10. SDS-Page Analysis
2.11. Liquid Chromatography-Tandem Mass-Spectrometry of Purified P1 Virions
2.12. Bioinformatic Analyses
2.13. Purification of Virions by CsCl Isopycnic Centrifugation
2.14. Purification of Virion Sub-Assembly by CsCl Step-Gradient Centrifugation
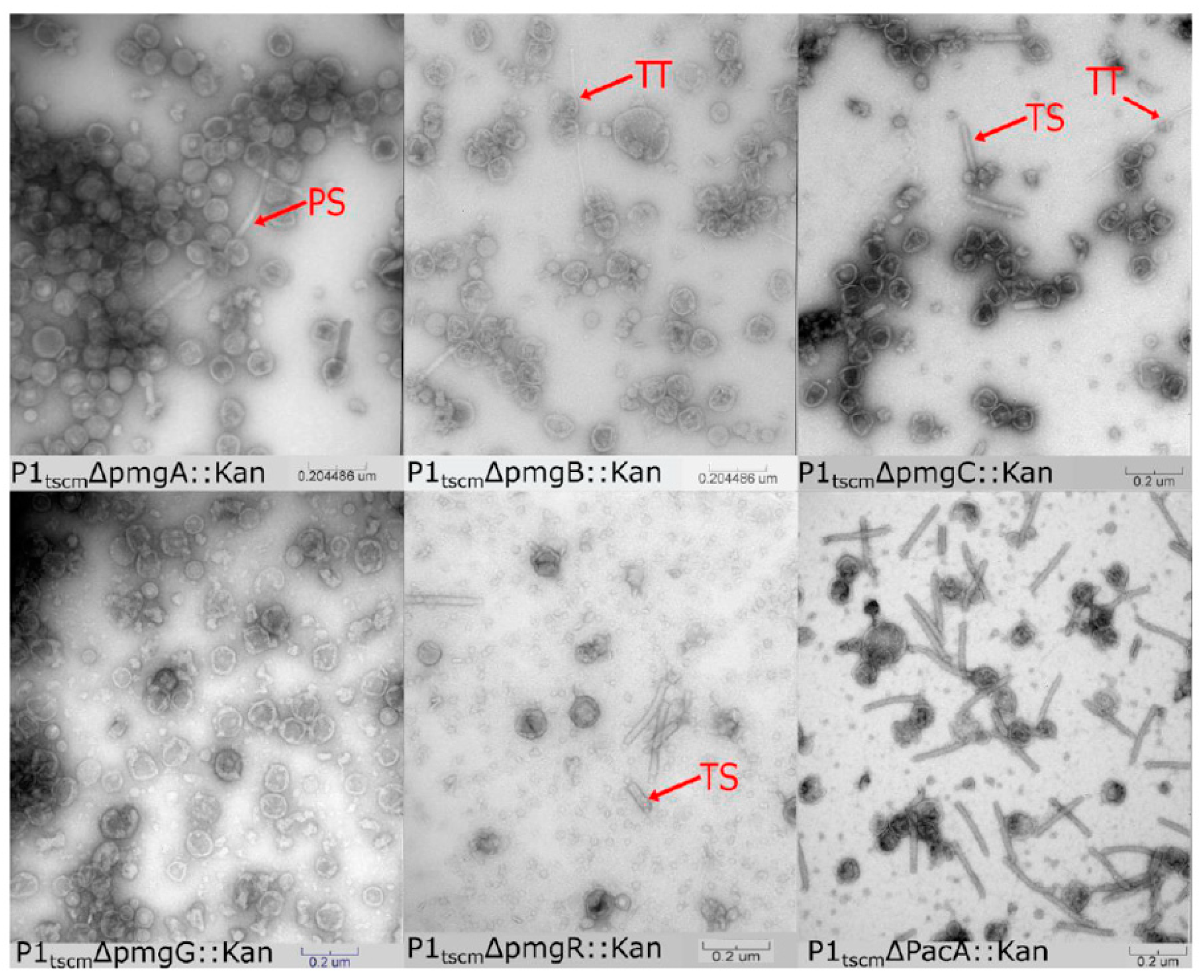
2.15. Complementation of P1 Δpmg Mutants
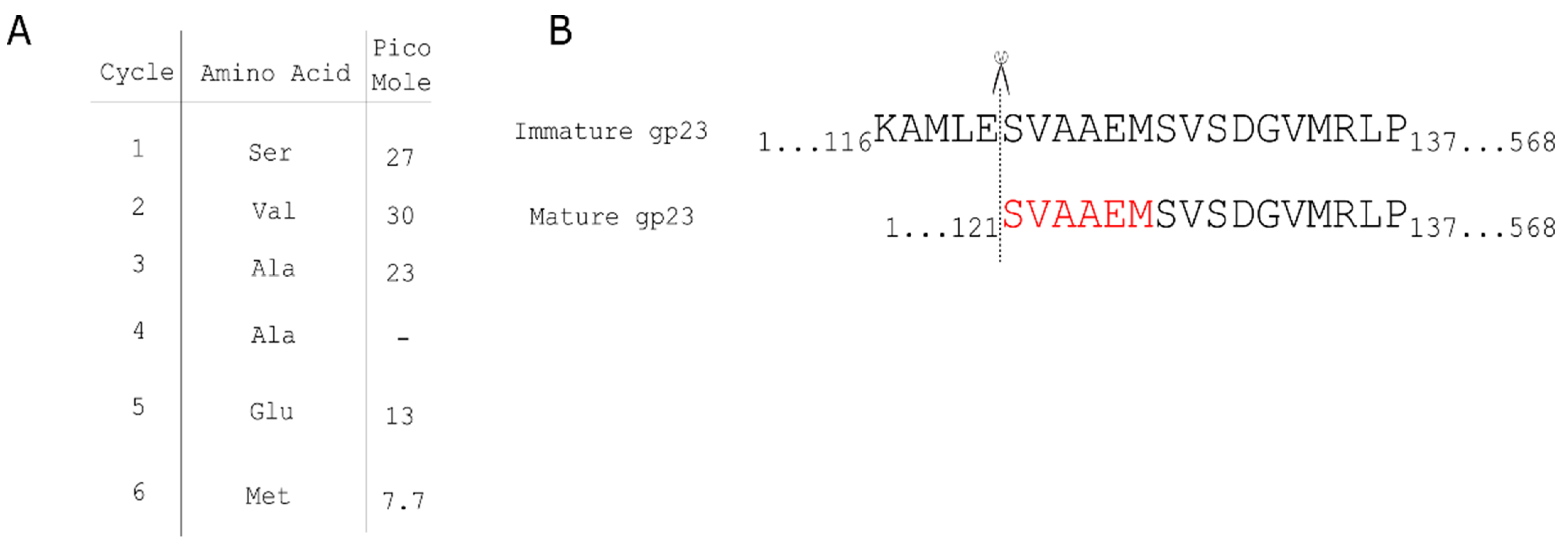
2.16. Edman Analysis
2.17. P1ΔdarB Transformation and pBAD24_N30-tetR-mCherry Induction
2.18. Fluorescence Microscopy Imaging
3. Results
3.1. Mass-Spectrometry Reveals the Proteome of the P1 Virion
3.1.1. P1 Head Proteins Identified by LC-MS/MS
3.1.2. P1 Tail Proteins Identified by LC-MS/MS
3.1.3. P1 Baseplate Proteins Identified by LC-MS/MS
3.2. Identification of Additional Morphogenetic Genes in P1
3.2.1. Assigning Roles to the Newly Identified Essential Genes in P1
3.2.2. Identification of Additional Structural Proteins
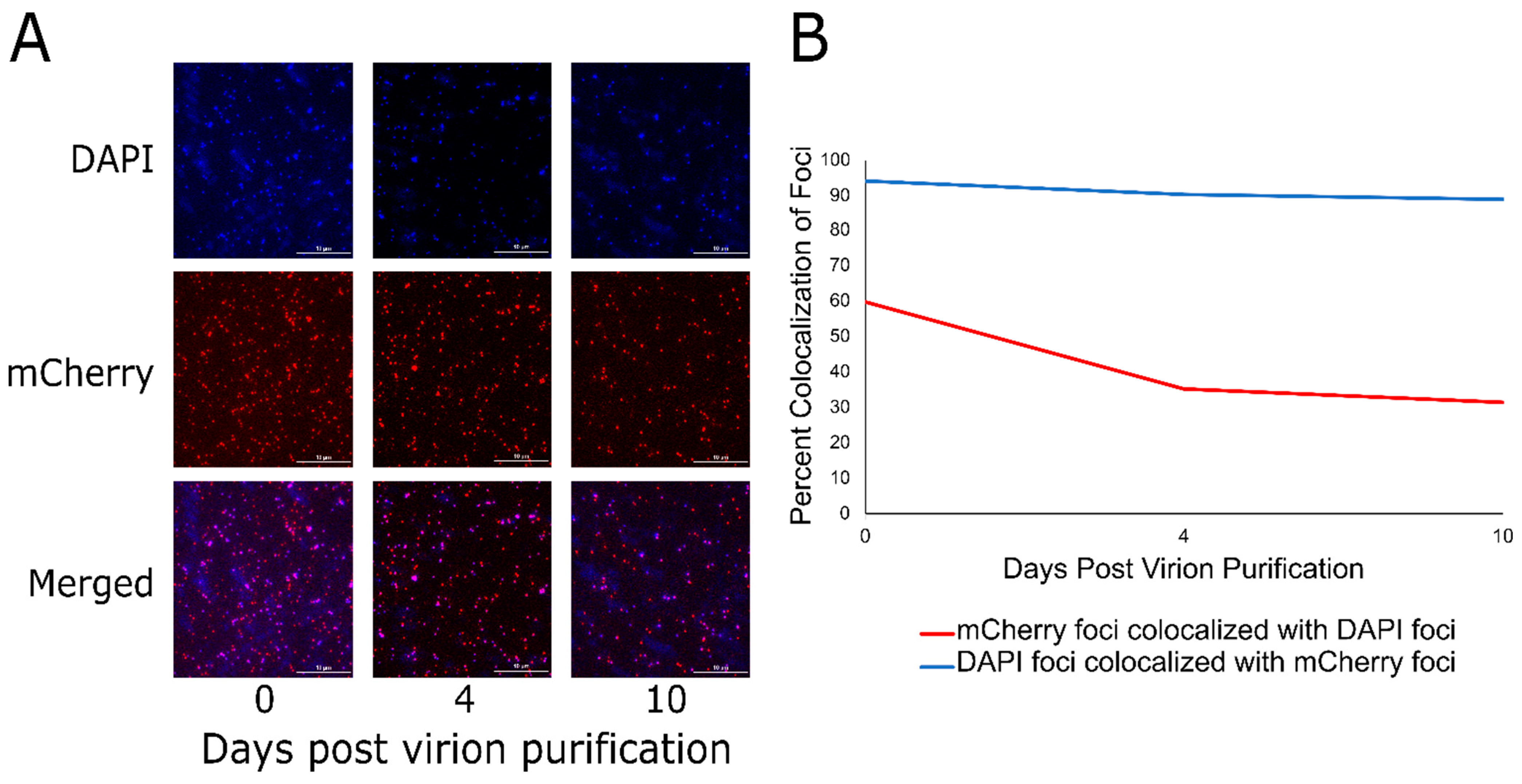
3.3. Localization of DarB into the Capsid
3.4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertani, G. Studies on lysogenesis I: The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lennox, E. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef]
- Yarmolinsky, M.B.; Sternberg, N. Bacteriophage P1. In The Bacteriophages; Calendar, R., Ed.; Springer: Boston, MA, USA, 1988; pp. 291–438. [Google Scholar]
- Arber, W.; Dussoix, D. Host specificity of DNA produced by Escherichia coli: I. Host controlled modification of bacteriophage λ. J. Mol. Biol. 1962, 5, 18–36. [Google Scholar] [CrossRef]
- Iida, S.; Streiff, M.B.; Bickle, T.A.; Arber, W. Two DNA antirestriction systems of bacteriophage P1, darA, and darB: Characterization of darA—Phages. Virology 1987, 157, 156–166. [Google Scholar] [CrossRef]
- Piya, D.; Vara, L.; Russell, W.K.; Young, R.; Gill, J.J. The multicomponent antirestriction system of phage P1 is linked to capsid morphogenesis. Mol. Microbiol. 2017, 105, 399–412. [Google Scholar] [CrossRef]
- Gilcrease, E.B.; Casjens, S.R. The genome sequence of Escherichia coli tailed phage D6 and the diversity of Enterobacteriales circular plasmid prophages. Virology 2018, 515, 203–214. [Google Scholar] [CrossRef]
- Bai, L.; Wang, J.; Hurley, D.; Yu, Z.; Wang, L.; Chen, Q.; Li, J.; Li, F.; Fanning, S. A novel disrupted mcr-1 gene and a lysogenized phage P1-like sequence detected from a large conjugative plasmid, cultured from a human atypical enteropathogenic Escherichia coli (aEPEC) recovered in China. J. Antimicrob. Chemother. 2017, 72, 1531–1533. [Google Scholar] [CrossRef][Green Version]
- Walker, J.T.; Walker, D.H. Coliphage P1 morphogenesis: Analysis of mutants by electron microscopy. J. Virol. 1983, 45, 1118–1139. [Google Scholar] [CrossRef]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [CrossRef]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Casjens, S.; Uetz, P. The protein interaction map of bacteriophage lambda. BMC Microbiol. 2011, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Valentine, R.C.; Shapiro, B.M.; Stadtman, E.R. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 1968, 7, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.J.; Summer, E.J.; Russell, W.K.; Cologna, S.M.; Carlile, T.M.; Fuller, A.C.; Kitsopoulos, K.; Mebane, L.M.; Parkinson, B.N.; Sullivan, D.; et al. Genomes and Characterization of Phages Bcep22 and BcepIL02, Founders of a Novel Phage Type in Burkholderia cenocepacia. J. Bacteriol. 2011, 193, 5300–5313. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Trinh, J.T.; McIntosh, C.S.; Christenson, B.; Balázsi, G.; Zeng, L. Lysis-lysogeny coexistence: Prophage integration during lytic development. MicrobiologyOpen 2016, 6, e00395. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Chait, B.T. Chemistry: Mass Spectrometry: Bottom-Up or Top-Down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, K.; Yang, X.; Sun, L.; You, J.; Pan, X.; Cui, X.; Yang, H. Characterization of a novel lytic podovirus O4 of Pseudomonas aeruginosa. Arch. Virol. 2018, 163, 2377–2383. [Google Scholar] [CrossRef] [PubMed]
- Keifer, D.Z.; Motwani, T.; Teschke, C.M.; Jarrold, M.F. Measurement of the accurate mass of a 50 MDa infectious virus. Rapid Commun. Mass Spectrom. 2016, 30, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M.; Sattelberger, E.; Inman, R.B.; Calendar, R.; Loessner, M.J. Genome and proteome of Listeria monocytogenes phage PSA: An unusual case for programmed + 1 translational frameshifting in structural protein synthesis. Mol. Microbiol. 2003, 50, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.D.; Martin, N.L.; Kropinski, A. The genome and proteome of coliphage T1. Virology 2004, 318, 245–266. [Google Scholar] [CrossRef]
- Walker, J.T.; Walker, D.H. Structural proteins of coliphage P1. Prog. Clin. Biol. Res. 1981, 64, 69–77. [Google Scholar]
- Taylor, N.M.I.; Prokhorov, N.; Guerrero-Ferreira, R.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef]
- Huang, R.K.; Khayat, R.; Lee, K.K.; Gertsman, I.; Duda, R.; Hendrix, R.W.; Johnson, J.E. The Prohead-I Structure of Bacteriophage HK97: Implications for Scaffold-Mediated Control of Particle Assembly and Maturation. J. Mol. Biol. 2011, 408, 541–554. [Google Scholar] [CrossRef]
- Effantin, G.; Boulanger, P.; Neumann, E.; Letellier, L.; Conway, J. Bacteriophage T5 Structure Reveals Similarities with HK97 and T4 Suggesting Evolutionary Relationships. J. Mol. Biol. 2006, 361, 993–1002. [Google Scholar] [CrossRef]
- Yap, M.L.; Rossmann, M.G. Structure and function of bacteriophage T4. Futur. Microbiol. 2014, 9, 1319–1327. [Google Scholar] [CrossRef]
- Hendrix, R.W. Bacteriophage HK97: Assembly of the Capsid and Evolutionary Connections. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2005; Volume 64, pp. 1–14. [Google Scholar] [CrossRef]
- Roos, W.H.; Gertsman, I.; May, E.R.; Brooks, C.L.; Johnson, J.E.; Wuite, G.J.L. Mechanics of bacteriophage maturation. Proc. Natl. Acad. Sci. USA 2012, 109, 2342–2347. [Google Scholar] [CrossRef]
- Streiff, M.B.; Iida, S.; Bickle, T.A. Expression and proteolytic processing of the darA antirestriction gene product of bacteriophage P1. Virology 1987, 157, 167–171. [Google Scholar] [CrossRef]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef] [PubMed]
- Duda, R.L.; Oh, B.; Hendrix, R.W. Functional Domains of the HK97 Capsid Maturation Protease and the Mechanisms of Protein Encapsidation. J. Mol. Biol. 2013, 425, 2765–2781. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W. Tail Length Determination in Double-Stranded DNA Bacteriophages. Curr. Top. Microbiol. Immunol. 1988, 136, 21–29. [Google Scholar] [CrossRef]
- Lehnherr, H.; Hansen, A.-M.; Ilyina, T. Penetration of the bacterial cell wall: A family of lytic transglycosylases in bacteriophages and conjugative plasmids. Mol. Microbiol. 1998, 30, 454–457. [Google Scholar] [CrossRef]
- Xu, J.; Hendrix, R.W.; Duda, R.L. Conserved Translational Frameshift in dsDNA Bacteriophage Tail Assembly Genes. Mol. Cell 2004, 16, 11–21. [Google Scholar] [CrossRef]
- Fokine, A.; Zhang, Z.; Kanamaru, S.; Bowman, V.D.; Aksyuk, A.A.; Arisaka, F.; Rao, V.B.; Rossmann, M.G. The Molecular Architecture of the Bacteriophage T4 Neck. J. Mol. Biol. 2013, 425, 1731–1744. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Navruzbekov, G.A.; Kurochkina, L.P.; Strelkov, S.V.; Mesyanzhinov, V.V.; Rossmann, M.G. The structure of bacteriophage T4 gene product 9: The trigger for tail contraction. Structure 1999, 7, 1213–1222. [Google Scholar] [CrossRef]
- Van Raaij, M.J.; Schoehn, G.; Burda, M.R.; Miller, S. Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J. Mol. Biol. 2001, 314, 1137–1146. [Google Scholar] [CrossRef]
- Abedon, S.T.; Yin, J. Bacteriophage Plaques: Theory and Analysis. Methods Mol. Biol. 2009, 501, 161–174. [Google Scholar] [CrossRef]
- Zhang, K.; Young, R.; Zeng, L. Bacteriophage P1 does not show spatial preference when infecting Escherichia coli. Virology 2020, 542, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Skorupski, K.; Pierce, J.C.; Sauer, B.; Sternberg, N.; Skorupski, K.; Pierce, J.C.; Sauer, B.; Sternberg, N. Bacteriophage P1 genes involved in the recognition and cleavage of the phage packaging site (pac). J. Mol. Biol. 1992, 223, 977–989. [Google Scholar] [CrossRef]
- Luftig, R.B.; Ganz, C. Bacteriophage T4 Head Morphogenesis IV. Comparison of Gene 16-, 17-, and 49-Defective Head Structures 2. J. Virol. 1972, 10, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Shneider, M.M.; Leiman, P.G.; Kuhn, A.; Kiefer, D. The Central Spike Complex of Bacteriophage T4 Contacts PpiD in the Periplasm of Escherichia coli. Viruses 2020, 12, 1135. [Google Scholar] [CrossRef]
- Bair, C.L.; Black, L.W. A Type IV Modification Dependent Restriction Nuclease that Targets Glucosylated Hydroxymethyl Cytosine Modified DNAs. J. Mol. Biol. 2007, 366, 768–778. [Google Scholar] [CrossRef]
- Mullaney, J.; Black, L.W. Capsid Targeting Sequence Targets Foreign Proteins into Bacteriophage T4 and Permits Proteolytic Processing. J. Mol. Biol. 1996, 261, 372–385. [Google Scholar] [CrossRef]
- Mullaney, J.M.; Black, L.W. Bacteriophage T4 Capsid Packaging and Unpackaging of DNA and Proteins. In Virus Hybrids as Nanomaterials: Methods and Protocols; Lin, B., Ratna, B., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 69–85. [Google Scholar]
- Kaiser, D.; Dworkin, M. Gene Transfer to Myxobacterium by Escherichia coli Phage P1. Science 1975, 187, 653–654. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Liu, J.; Chen, C.-Y.; Shiomi, D.; Niki, H.; Margolin, W. Visualization of bacteriophage P1 infection by cryo-electron tomography of tiny Escherichia coli. Virology 2011, 417, 304–311. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Van Raaij, M.J.; Leiman, P.G. Contractile injection systems of bacteriophages and related systems. Mol. Microbiol. 2018, 108, 6–15. [Google Scholar] [CrossRef]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, P.; Jacquot, P.; Plançon, L.; Chami, M.; Engel, A.; Parquet, C.; Herbeuval, C.; Letellier, L. Phage T5 Straight Tail Fiber Is a Multifunctional Protein Acting as a Tape Measure and Carrying Fusogenic and Muralytic Activities. J. Biol. Chem. 2008, 283, 13556–13564. [Google Scholar] [CrossRef] [PubMed]
- Piuri, M.; Hatfull, G.F. A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol. Microbiol. 2006, 62, 1569–1585. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
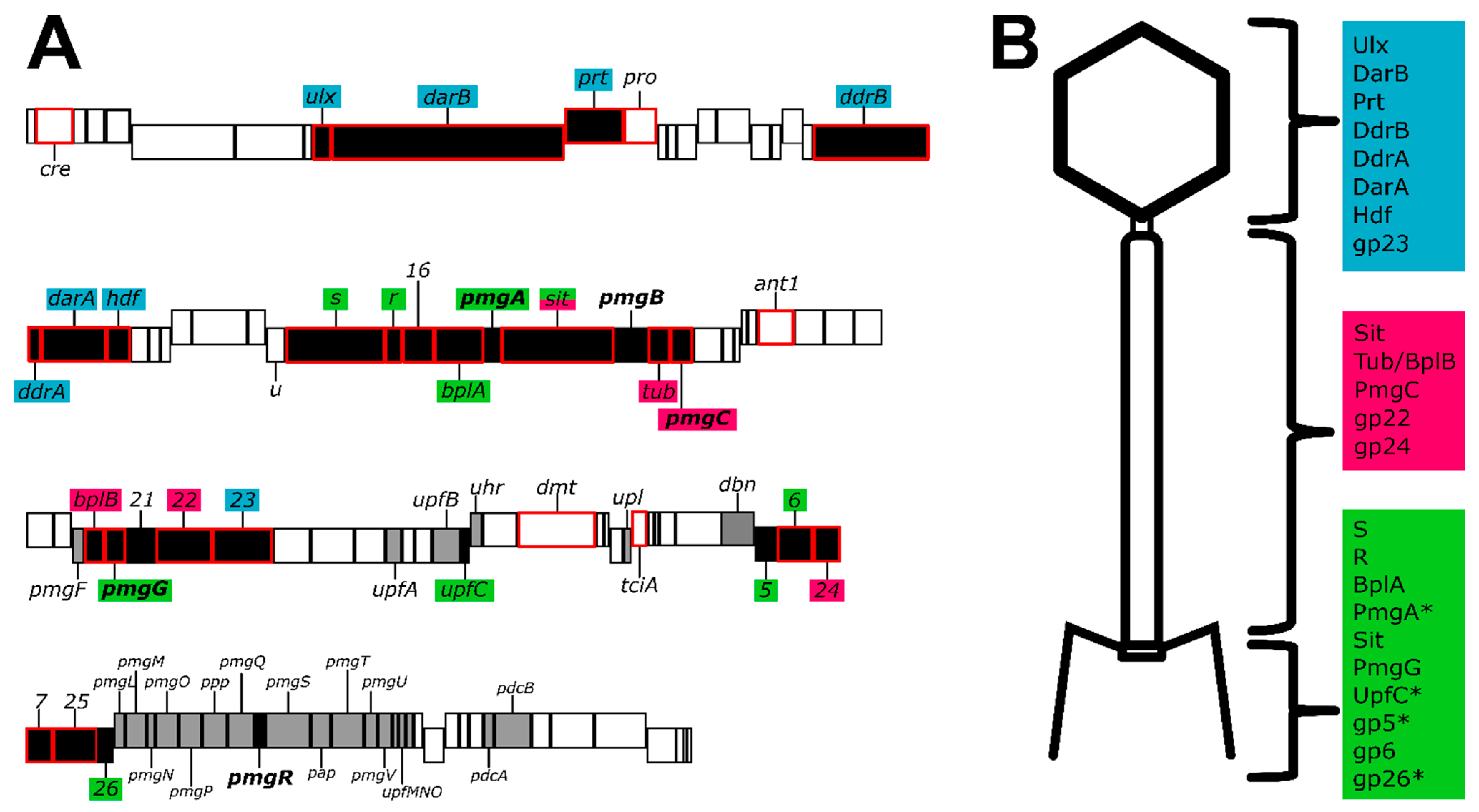{kind=link}
| Detected Protein | Predicted Function | Molecular Mass (kDa) | Number of Unique Peptides | Coverage (%) | Predicted Location of Protein | ORF Number | Source | GenBank ID |
|---|---|---|---|---|---|---|---|---|
| Ulx | Aids DarB localization | 17 | 3 | 20.5 | Head | 9 | [6] | 2777418 |
| DarB | Inhibition of EcoK and EcoB endonucleases | 251.4 | 37 | 25.8 | Head | 10 | [6] | 2777481 |
| Prt | Portal protein | 62.7 | 11 | 37.7 | Head | 11 | 2777482 | |
| Pro | Prohead protease | 36.8 | 3 | 9.7 | Head † | 12 | 2777381 | |
| DdrB | Aids DarB addition to capsid | 108.7 | 39 | 59.5 | Head | 22 | [6] | 2777413 |
| DdrA | Antagonist of EcoA endonuclease | 13 | 2 | 27.2 | Head | 23 | [6] | 2777414 |
| DarA | Head size determinant and Dar system incorporation | 69.4 | 27 | 67.4 | Head | 24 | [6] | 2777415 |
| Hdf | Head size determinant and Dar system incorporation | 22.1 | 7 | 39.9 | Head | 25 | [6] | 2777469 |
| S | Tail fiber specificity | 104.8 | 18 | 32.8 | Baseplate | 33 | [10] | 2777437 |
| R | Baseplate wedge | 15.9 | 3 | 26.3 | Baseplate | 34 | This work | 2777404 |
| gp16 | Baseplate structure * | 31.3 | 2 | 11.5 | Baseplate †,* | 35 | 2777405 | |
| BplA | Baseplate wedge | 53.5 | 7 | 21.5 | Baseplate | 36 | This work | 2777406 |
| Sit | Tape measure protein | 120.6 | 23 | 27.8 | Tail & Baseplate | 38 | This work | 2777408 |
| Tub | Tail tube a | 22.3 | 6 | 44.8 | Tail | 40 | This work | 2777479 |
| PmgC | Tail adaptor | 31.9 | 4 | 31 | Tail | 41 | This work | 2777480 |
| BplB | Tail tube a | 18.7 | 7 | 33.1 | Tail | 54 | This work | 2777444 |
| PmgG | Baseplate-tail tube junction | 20.5 | 2 | 20.2 | Baseplate | 55 | This work | 2777384 |
| gp22 | Tail sheath protein | 56.9 | 22 | 56.1 | Tail | 57 | [10] | 2777386 |
| gp23 | Major capsid protein | 62.2 | 24 | 47.5 | Head | 58 | [10] | 2777387 |
| gp6 | Baseplate hub | 37.2 | 4 | 16.8 | Baseplate | 83 | This work | 2777424 |
| gp24 | Tail terminator | 28.9 | 5 | 22.2 | Tail | 84 | This work | 2777425 |
| gp7 | Tail Stability * | 27.1 | 2 | 11.1 | Tail †,* | 85 | [10] | 2777426 |
| gp25 | Tail Stability * | 45.8 | 2 | 8.4 | Tail †,* | 86 | [10] | 2777427 |
| Lysate | Lysogens Established/mL Induced lysate | Fold Increase in Lysogen Establishment | Estimated Virions/mL in Induced Lysate |
|---|---|---|---|
| P1ΔpmgA | 1.8 × 101 | - | 6.0 × 102 |
| P1ΔpmgA + ppmgA | 2.0 × 102 | 11 | 6.7 × 103 |
| P1ΔpmgB | 1.5 × 101 | - | 5.0 × 102 |
| P1ΔpmgB + ppmgB | 7.0 × 101 | 4.7 | 2.3 × 102 |
| P1ΔpmgC | 2.2 × 101 | - | 7.3 × 102 |
| P1ΔpmgC + ppmgC | 1.3 × 102 | 6.1 | 4.4 × 103 |
| P1ΔpmgG | 3.4 × 102 | - | 1.1 × 104 |
| P1ΔpmgG + ppmgG | 1.6 × 105 | 460 | 5.3 × 106 |
| P1ΔpmgR | 1.3 × 101 | - | 4.3 × 102 |
| P1ΔpmgR + ppmgR | 2.4 × 103 | 180 | 8.0 × 104 |
| P1 | 3.0 × 105 | - | 1.0 × 107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzales, M.F.; Piya, D.K.; Koehler, B.; Zhang, K.; Yu, Z.; Zeng, L.; Gill, J.J. New Insights into the Structure and Assembly of Bacteriophage P1. Viruses 2022, 14, 678. https://doi.org/10.3390/v14040678
Gonzales MF, Piya DK, Koehler B, Zhang K, Yu Z, Zeng L, Gill JJ. New Insights into the Structure and Assembly of Bacteriophage P1. Viruses. 2022; 14(4):678. https://doi.org/10.3390/v14040678
Chicago/Turabian StyleGonzales, Miguel F., Denish K. Piya, Brian Koehler, Kailun Zhang, Zihao Yu, Lanying Zeng, and Jason J. Gill. 2022. "New Insights into the Structure and Assembly of Bacteriophage P1" Viruses 14, no. 4: 678. https://doi.org/10.3390/v14040678
APA StyleGonzales, M. F., Piya, D. K., Koehler, B., Zhang, K., Yu, Z., Zeng, L., & Gill, J. J. (2022). New Insights into the Structure and Assembly of Bacteriophage P1. Viruses, 14(4), 678. https://doi.org/10.3390/v14040678