
Imaging of Hepatitis B Virus Nucleic Acids: Current Advances and Challenges

 , , , and
, , , and
Abstract
1. Problems and Significance of cccDNA and mRNA Visualisation
2. Techniques Applied for HBV Nucleic Acid Visualisation
2.1. Electron Microscopy
2.2. In Situ Hybridisation
3. Principal Findings on HBV by Nucleic Acid Visualisation
3.1. Tissue Samples—Imaging with Low Resolution
3.2. Single Cell Analyses—Towards High-Resolution Imaging
4. Recent Developments in DNA and mRNA Detection for Living Cells
4.1. CRISPR-Based Detection
4.2. Non-CRISPR-Based Techniques
4.2.1. RNA Imaging
4.2.2. dsDNA Imaging
4.2.3. Alternative Fluorophores
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seto, W.K.; Lo, Y.R.; Pawlotsky, J.M.; Yuen, M.F. Chronic hepatitis B virus infection. Lancet 2018, 392, 2313–2324. [Google Scholar] [CrossRef]
- EASL, EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere with Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef]
- Peng, W.C.; Kraaier, L.J.; Kluiver, T.A. Hepatocyte organoids and cell transplantation: What the future holds. Exp. Mol. Med. 2021, 53, 1512–1528. [Google Scholar] [CrossRef]
- Wieland, S.F.; Spangenberg, H.C.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. USA 2004, 101, 2129–2134. [Google Scholar] [CrossRef]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef]
- Buti, M.; Tsai, N.; Petersen, J.; Flisiak, R.; Gurel, S.; Krastev, Z.; Aguilar Schall, R.; Flaherty, J.F.; Martins, E.B.; Charuworn, P.; et al. Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection. Dig. Dis. Sci. 2015, 60, 1457–1464. [Google Scholar] [CrossRef]
- Zhao, S.S.; Tang, L.H.; Dai, X.H.; Wang, W.; Zhou, R.R.; Chen, L.Z.; Fan, X.G. Comparison of the efficacy of tenofovir and adefovir in the treatment of chronic hepatitis B: A systematic review. Virol. J. 2011, 8, 111. [Google Scholar] [CrossRef]
- Ribeiro, R.M.; Germanidis, G.; Powers, K.A.; Pellegrin, B.; Nikolaidis, P.; Perelson, A.S.; Pawlotsky, J.M. Hepatitis B virus kinetics under antiviral therapy sheds light on differences in hepatitis B e antigen positive and negative infections. J. Infect. Dis. 2010, 202, 1309–1318. [Google Scholar] [CrossRef]
- Wang, X.; Le, N.; Denoth-Lippuner, A.; Barral, Y.; Kroschewski, R. Asymmetric partitioning of transfected DNA during mammalian cell division. Proc. Natl. Acad. Sci. USA 2016, 113, 7177–7182. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Volz, T.; Giersch, K.; Kah, J.; Raffa, G.; Petersen, J.; Lohse, A.W.; Beninati, C.; Pollicino, T.; Urban, S.; et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 2018, 67, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Lythgoe, K.A.; Lumley, S.F.; Pellis, L.; McKeating, J.A.; Matthews, P.C. Estimating hepatitis B virus cccDNA persistence in chronic infection. Virus Evol. 2021, 7, veaa063. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, B.; Cai, D.; Zong, Y.; Wu, Y.; Liu, S.; Mercier, A.; Guo, H.; Hou, J.; Colonno, R.; et al. Rapid Turnover of Hepatitis B Virus Covalently Closed Circular DNA Indicated by Monitoring Emergence and Reversion of Signature-Mutation in Treated Chronic Hepatitis B Patients. Hepatology 2021, 73, 41–52. [Google Scholar] [CrossRef]
- Mason, W.S.; Xu, C.; Low, H.C.; Saputelli, J.; Aldrich, C.E.; Scougall, C.; Grosse, A.; Colonno, R.; Litwin, S.; Jilbert, A.R. The amount of hepatocyte turnover that occurred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J. Virol. 2009, 83, 1778–1789. [Google Scholar] [CrossRef]
- Murray, J.M.; Wieland, S.F.; Purcell, R.H.; Chisari, F.V. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 2005, 102, 17780–17785. [Google Scholar] [CrossRef]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Balagopal, A.; Grudda, T.; Ribeiro, R.M.; Saad, Y.S.; Hwang, H.S.; Quinn, J.; Murphy, M.; Ward, K.; Sterling, R.K.; Zhang, Y.; et al. Single hepatocytes show persistence and transcriptional inactivity of hepatitis B. JCI Insight 2020, 5, e140584. [Google Scholar] [CrossRef]
- Chauhan, R.; Michalak, T.I. Earliest hepatitis B virus-hepatocyte genome integration: Sites, mechanism, and significance in carcinogenesis. Hepatoma Res. 2021, 7, 20. [Google Scholar] [CrossRef]
- Lindh, M.; Rydell, G.E.; Larsson, S.B. Impact of integrated viral DNA on the goal to clear hepatitis B surface antigen with different therapeutic strategies. Curr. Opin. Virol. 2018, 30, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Kordon, M.M.; Zarębski, M.; Solarczyk, K.; Ma, H.; Pederson, T.; Dobrucki, J.W. STRIDE—A fluorescence method for direct, specific in situ detection of individual single- or double-strand DNA breaks in fixed cells. Nucleic Acids Res. 2020, 48, e14. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.-H.; Chen, D.-Y.; Ye, L.-P.; Sheng, G.; Gong, J.-J.; Chen, B.-H.; Lu, Y.-M.; Han, F. CRISPR-Sunspot: Imaging of endogenous low-abundance RNA at the single-molecule level in live cells. Theranostics 2020, 10, 10993–11012. [Google Scholar] [CrossRef]
- Rydell, G.E.; Prakash, K.; Larsson, S.B.; Skoglund, C.; Ringlander, J.; Andersson, M.; Castedal, M.; Norder, H.; Lindh, M. Quantification of viral RNA in multiple pieces of explant liver tissue shows distinct focal differences of hepatitis B infection. J. Infect. Dis. 2021, jiab469. [Google Scholar] [CrossRef]
- Le, C.; Liu, Y.; Lopez-Orozco, J.; Joyce, M.A.; Le, X.C.; Tyrrell, D.L. CRISPR Technique Incorporated with Single-Cell RNA Sequencing for Studying Hepatitis B Infection. Anal. Chem. 2021, 93, 10756–10761. [Google Scholar] [CrossRef]
- Kremsdorf, D.; Lekbaby, B.; Bablon, P.; Sotty, J.; Augustin, J.; Schnuriger, A.; Pol, J.; Soussan, P. Alternative splicing of viral transcripts: The dark side of HBV. Gut 2021, 70, 2373–2382. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, S.; Li, L.; Huang, X.; Xing, S.; Zhang, Y.; Qiu, G.; Han, Z.; Shang, Y.; Sun, D.E.; et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Gowans, E.J.; Burrell, C.J.; Jilbert, A.R.; Marmion, B.P. Detection of hepatitis B virus DNA sequences in infected hepatocytes by in situ cytohybridisation. J. Med. Virol. 1981, 8, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Burrell, C.J.; Gowans, E.J.; Rowland, R.; Hall, P.; Jilbert, A.R.; Marmion, B.P. Correlation between liver histology and markers of hepatitis B virus replication in infected patients: A study by in situ hybridization. Hepatology 1984, 4, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Blum, H.E.; Stowring, L.; Figus, A.; Montgomery, C.K.; Haase, A.T.; Vyas, G.N. Detection of hepatitis B virus DNA in hepatocytes, bile duct epithelium, and vascular elements by in situ hybridization. Proc. Natl. Acad. Sci. USA 1983, 80, 6685–6688. [Google Scholar] [CrossRef] [PubMed]
- Gowans, E.J.; Burrell, C.J.; Jilbert, A.R.; Marmion, B.P. Patterns of single- and double-stranded hepatitis B virus DNA and viral antigen accumulation in infected liver cells. J. Gen. Virol. 1983, 64 Pt 6, 1229–1239. [Google Scholar] [CrossRef]
- Blum, H.E.; Haase, A.T.; Harris, J.D.; Walker, D.; Vyas, G.N. Asymmetric replication of hepatitis B virus DNA in human liver: Demonstration of cytoplasmic minus-strand DNA by blot analyses and in situ hybridization. Virology 1984, 139, 87–96. [Google Scholar] [CrossRef]
- Rijntjes, P.J.; Van Ditzhuijsen, T.J.; Van Loon, A.M.; Van Haelst, U.J.; Bronkhorst, F.B.; Yap, S.H. Hepatitis B virus DNA detected in formalin-fixed liver specimens and its relation to serologic markers and histopathologic features in chronic liver disease. Am. J. Pathol. 1985, 120, 411–418. [Google Scholar]
- Negro, F.; Berninger, M.; Chiaberge, E.; Gugliotta, P.; Bussolati, G.; Actis, G.C.; Rizzetto, M.; Bonino, F. Detection of HBV-DNA by in situ hybridization using a biotin-labeled probe. J. Med. Virol. 1985, 15, 373–382. [Google Scholar] [CrossRef]
- Naoumov, N.V.; Daniels, H.M.; Davison, F.; Eddleston, A.L.; Alexander, G.J.; Williams, R. Identification of hepatitis B virus-DNA in the liver by in situ hybridization using a biotinylated probe. Relation to HBcAg expression and histology. J. Hepatol. 1993, 19, 204–210. [Google Scholar] [CrossRef]
- Han, K.H.; Hollinger, F.B.; Noonan, C.A.; Yoffe, B. Simultaneous detection of HBV-specific antigens and DNA in paraffin-embedded liver tissue by immunohistochemistry and in situ hybridization using a digoxigenin-labeled probe. J. Virol. Methods 1992, 37, 89–97. [Google Scholar] [CrossRef]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell Rep. 2021, 35, 109288. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, Y.; Li, G.; Shen, C.; Meng, Z.; Zheng, J.; Jia, Y.; Chen, S.; Zhang, X.; Zhu, M.; et al. Relationship between serum HBV-RNA levels and intrahepatic viral as well as histologic activity markers in entecavir-treated patients. J. Hepatol. 2017, 68, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef]
- Stone, D.; Long, K.R.; Loprieno, M.A.; De Silva Feelixge, H.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther. Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, W.; Zheng, Y.; Wang, W.; Bai, L.; Chen, L.; Feng, Y.; Zhang, Z.; Yuan, Z. In situ analysis of intrahepatic virological events in chronic hepatitis B virus infection. J. Clin. Investig. 2016, 126, 1079–1092. [Google Scholar] [CrossRef]
- Liu, D.; Xu, T.; Shi, B.; Lu, W.; Zheng, Y.; Feng, Y.; Yuan, Z.; Zhang, X.; Zhang, Z. Clinical relevance of the in situ assay for HBV DNA: A cross-sectional study in patients with chronic hepatitis B. J. Clin. Pathol. 2020, 73, 813–818. [Google Scholar] [CrossRef]
- Li, C.; Zhang, W.; Shi, B.; Chen, G.; Zheng, Y.; An, Y.; Sun, M.; Feng, Y.; Shang, Q.; Zhang, X. Evaluation of the in situ assay for HBV DNA: An observational real-world study in chronic hepatitis B. Medicine 2021, 100, e27220. [Google Scholar] [CrossRef]
- Zhang, X.; Yue, L.; Zhang, Z.; Yuan, Z. Establishment of a fluorescent in situ hybridization assay for imaging hepatitis B virus nucleic acids in cell culture models. Emerg. Microbes Infect. 2017, 6, e98. [Google Scholar] [CrossRef]
- Yue, L.; Li, C.; Xu, M.; Wu, M.; Ding, J.; Liu, J.; Zhang, X.; Yuan, Z. Probing the spatiotemporal patterns of HBV multiplication reveals novel features of its subcellular processes. PLoS Pathog. 2021, 17, e1009838. [Google Scholar] [CrossRef]
- Wieland, S.F.; Asabe, S.; Engle, R.E.; Purcell, R.H.; Chisari, F.V. Limited hepatitis B virus replication space in the chronically hepatitis C virus-infected liver. J. Virol. 2014, 88, 5184–5188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaudhary, N.; Nho, S.-H.; Cho, H.; Gantumur, N.; Ra, J.S.; Myung, K.; Kim, H. Background-suppressed live visualization of genomic loci with an improved CRISPR system based on a split fluorophore. Genome Res. 2020, 30, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Urbanek, M.O.; Galka-Marciniak, P.; Olejniczak, M.; Krzyzosiak, W.J. RNA imaging in living cells—Methods and applications. RNA biology 2014, 11, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, J. RNA Fluorescence with Light-Up Aptamers. Front. Chem. 2016, 4, 29. [Google Scholar] [CrossRef]
- Cawte, A.D.; Unrau, P.J.; Rueda, D.S. Live cell imaging of single RNA molecules with fluorogenic Mango II arrays. Nat. Commun. 2020, 11, 1283. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Ying, Y.; Wu, R.; Chen, A.K. Recent Advances in the Molecular Beacon Technology for Live-Cell Single-Molecule Imaging. iScience 2020, 23, 101801. [Google Scholar] [CrossRef] [PubMed]
- Bann, D.V.; Parent, L.J. Application of live-cell RNA imaging techniques to the study of retroviral RNA trafficking. Viruses 2012, 4, 963–979. [Google Scholar] [CrossRef]
- Wang, Z.G.; Liu, S.L.; Pang, D.W. Quantum Dots: A Promising Fluorescent Label for Probing Virus Trafficking. Acc. Chem. Res. 2021, 54, 2991–3002. [Google Scholar] [CrossRef]
- Barroso, M.M. Quantum dots in cell biology. J. Histochem. Cytochem. 2011, 59, 237–251. [Google Scholar] [CrossRef]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Vilaivan, T. Fluorogenic PNA probes. Beilstein J. Org. Chem. 2018, 14, 253–281. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Demidov, V.V.; Gildea, B.D.; Fiandaca, M.J.; Coull, J.C.; Frank-Kamenetskii, M.D. PNA beacons for duplex DNA. Antisense Nucleic Acid Drug Dev. 2001, 11, 265–270. [Google Scholar] [CrossRef]
- Demidov, V.V.; Frank-Kamenetskii, M.D. PNA openers and their applications. Methods Mol. Biol. 2002, 208, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Cassany, A.; Ragues, J.; Guan, T.; Begu, D.; Wodrich, H.; Kann, M.; Nemerow, G.R.; Gerace, L. Nuclear import of adenovirus DNA involves direct interaction of hexon with an N-terminal domain of the nucleoporin Nup214. J. Virol. 2015, 89, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Gowans, E.J.; Burrell, C.J.; Jilbert, A.R.; Marmion, B.P. Cytoplasmic (but not nuclear) hepatitis B virus (HBV) core antigen reflects HBV DNA synthesis at the level of the infected hepatocyte. Intervirology 1985, 24, 220–225. [Google Scholar] [CrossRef]
- Blum, H.E.; Haase, A.T.; Vyas, G.N. Molecular pathogenesis of hepatitis B virus infection: Simultaneous detection of viral DNA and antigens in paraffin-embedded liver sections. Lancet 1984, 2, 771–775. [Google Scholar] [CrossRef]
- Negro, F.; Pacchioni, D.; Mondardini, A.; Bussolati, G.; Bonino, F. In situ hybridization in viral hepatitis. Liver 1992, 12, 217–226. [Google Scholar] [CrossRef]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B—Like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Niedobitek, G.; Finn, T.; Herbst, H.; Stein, H. Detection of viral genomes in the liver by in situ hybridization using 35S-, bromodeoxyuridine-, and biotin-labeled probes. Am. J. Pathol. 1989, 134, 633–639. [Google Scholar] [PubMed]
- Revill, P.A.; Locarnini, S.A. New perspectives on the hepatitis B virus life cycle in the human liver. J. Clin. Investig. 2016, 126, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Heimstadt, O. Das Fluoreszenzmikroskop. Z. Wiss Mikrosk. 1911, 28, 330. [Google Scholar]
- Reichert, K. Das Fluoreszenzmikroskop. Phys. Z 1911, 12, 1010. [Google Scholar]
- Bauman, J.G.; Wiegant, J.; Borst, P.; van Duijn, P. A new method for fluorescence microscopical localisation of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. Exp. Cell Res. 1980, 128, 485–490. [Google Scholar] [CrossRef]
- Langer-Safer, P.R.; Levine, M.; Ward, D.C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. USA 1982, 79, 4381–4385. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Cheng, X.; Li, Y.; Valdez, K.; Chen, W.; Liang, T.J. Hepatitis B Virus Deregulates the Cell Cycle to Promote Viral Replication and a Premalignant Phenotype. J. Virol. 2018, 92, e00722-18. [Google Scholar] [CrossRef]
- Allweiss, L.; Giersch, K.; Pirosu, A.; Volz, T.; Muench, R.C.; Beran, R.K.; Urban, S.; Javanbakht, H.; Fletcher, S.P.; Lutgehetmann, M.; et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut 2021, 71, 372–381. [Google Scholar] [CrossRef]
- Hensel, K.O.; Cantner, F.; Bangert, F.; Wirth, S.; Postberg, J. Episomal HBV persistence within transcribed host nuclear chromatin compartments involves HBx. Epigenet. Chromatin 2018, 11, 34. [Google Scholar] [CrossRef]
- Li, M.; Sohn, J.A.; Seeger, C. Distribution of Hepatitis B Virus Nuclear DNA. J. Virol. 2018, 92, e01391-17. [Google Scholar] [CrossRef]
- Summers, J.; Smith, P.M.; Horwich, A.L. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990, 64, 2819–2824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef] [PubMed]
- Stadelmayer, B.; Diederichs, A.; Chapus, F.; Rivoire, M.; Neveu, G.; Alam, A.; Fraisse, L.; Carter, K.; Testoni, B.; Zoulim, F. Full-length 5′RACE identifies all major HBV transcripts in HBV-infected hepatocytes and patient serum. J. Hepatol. 2020, 73, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Altinel, K.; Hashimoto, K.; Wei, Y.; Neuveut, C.; Gupta, I.; Suzuki, A.M.; Dos Santos, A.; Moreau, P.; Xia, T.; Kojima, S.; et al. Single-Nucleotide Resolution Mapping of Hepatitis B Virus Promoters in Infected Human Livers and Hepatocellular Carcinoma. J. Virol. 2016, 90, 10811–10822. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Riegler, J.; Wu, J.; Yen, T.S. Novel short transcripts of hepatitis B virus X gene derived from intragenic promoter. J. Biol. Chem. 1994, 269, 22593–22598. [Google Scholar] [CrossRef]
- Prakash, K.; Larsson, S.B.; Rydell, G.E.; Andersson, M.E.; Ringlander, J.; Norkrans, G.; Norder, H.; Lindh, M. Hepatitis B Virus RNA Profiles in Liver Biopsies by Digital Polymerase Chain Reaction. Hepatol. Commun. 2020, 4, 973–982. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Anton, T.; Karg, E.; Bultmann, S. Applications of the CRISPR/Cas system beyond gene editing. Biol. Methods Protocols 2018, 3, bpy002. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, K.; Li, J. Application of CRISPR/Cas9 Technology to HBV. Int. J. Mol. Sci. 2015, 16, 26077–26086. [Google Scholar] [CrossRef]
- Wu, X.; Mao, S.; Ying, Y.; Krueger, C.J.; Chen, A.K. Progress and Challenges for Live-cell Imaging of Genomic Loci Using CRISPR-based Platforms. Genom. Proteom. Bioinf. 2019, 17, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Peng, R.; Zhang, R.; Li, J. The applications of CRISPR/Cas system in molecular detection. J. Cell. Mol. Med. 2018, 22, 5807–5815. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [Google Scholar] [CrossRef]
- Qin, P.; Parlak, M.; Kuscu, C.; Bandaria, J.; Mir, M.; Szlachta, K.; Singh, R.; Darzacq, X.; Yildiz, A.; Adli, M. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017, 8, 14725. [Google Scholar] [CrossRef]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.-C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.-C.; Naseri, A.; Chung, Y.-C.; Grunwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA Scaffolds for Signal Amplification in Genome Imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef]
- Yang, L.-Z.; Wang, Y.; Li, S.-Q.; Yao, R.-W.; Luan, P.-F.; Wu, H.; Carmichael, G.G.; Chen, L.-L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997. [Google Scholar] [CrossRef]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Nilaratanakul, V.; Hauer, D.A.; Griffin, D.E. Development and characterization of Sindbis virus with encoded fluorescent RNA aptamer Spinach2 for imaging of replication and immune-mediated changes in intracellular viral RNA. J. Gen. Virol. 2017, 98, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Burch, B.D.; Garrido, C.; Margolis, D.M. Detection of human immunodeficiency virus RNAs in living cells using Spinach RNA aptamers. Virus Res. 2017, 228, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, P.; Nitin, N.; LaConte, L.; Woolums, A.; Bao, G. Live-cell characterization and analysis of a clinical isolate of bovine respiratory syncytial virus, using molecular beacons. J. Virol. 2006, 80, 682–688. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Wang, Z.-G.; Hu, Y.; Shi, X.-H.; Chen, H.-J.; Li, X.; Liu, S.-L.; Pang, D.-W. In-situ quantitation of genome release of Japanese encephalitis viruses by quantum dot-based single-virus tracking. Nano Today 2021, 40, 101271. [Google Scholar] [CrossRef]
- Jayagopal, A.; Halfpenny, K.C.; Perez, J.W.; Wright, D.W. Hairpin DNA-Functionalized Gold Colloids for the Imaging of mRNA in Live Cells. J. Am. Chem. Soc. 2010, 132, 9789–9796. [Google Scholar] [CrossRef]
- Wang, A.; Salazar, A.M.; Yates, M.V.; Mulchandani, A.; Chen, W. Visualization and detection of infectious coxsackievirus replication using a combined cell culture-molecular beacon assay. Appl Environ. Microbiol. 2005, 71, 8397–8401. [Google Scholar] [CrossRef][Green Version]
- Yeh, H.-Y.; Yates, M.V.; Mulchandani, A.; Chen, W. Visualizing the dynamics of viral replication in living cells via Tat peptide delivery of nuclease-resistant molecular beacons. Proc. Natl. Acad. Sci. USA 2008, 105, 17522. [Google Scholar] [CrossRef]
- Cui, Z.-Q.; Zhang, Z.-P.; Zhang, X.-E.; Wen, J.-K.; Zhou, Y.-F.; Xie, W.-H. Visualizing the dynamic behavior of poliovirus plus-strand RNA in living host cells. Nucleic Acids Res. 2005, 33, 3245–3252. [Google Scholar] [CrossRef]
- Wang, W.; Cui, Z.Q.; Han, H.; Zhang, Z.P.; Wei, H.P.; Zhou, Y.F.; Chen, Z.; Zhang, X.E. Imaging and characterizing influenza A virus mRNA transport in living cells. Nucleic Acids Res. 2008, 36, 4913–4928. [Google Scholar] [CrossRef]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94, e00135-20. [Google Scholar] [CrossRef] [PubMed]
- Mariamé, B.; Kappler-Gratias, S.; Kappler, M.; Balor, S.; Gallardo, F.; Bystricky, K. Real-Time Visualization and Quantification of Human Cytomegalovirus Replication in Living Cells Using the ANCHOR DNA Labeling Technology. J. Virol. 2018, 92, e00571-18. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In Vivo Labelling of Adenovirus DNA Identifies Chromatin Anchoring and Biphasic Genome Replication. J. Virol. 2018, 92, e00795-18. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, F.; Schmitt, D.; Brandely, R.; Brua, C.; Silvestre, N.; Findeli, A.; Foloppe, J.; Top, S.; Kappler-Gratias, S.; Quentin-Froignant, C.; et al. Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators. Biomedicines 2020, 8, 543. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, C.; Wiesmeijer, K.; Verwoerd, N.P.; Khazen, S.; Eils, R.; Tanke, H.J.; Dirks, R.W. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003, 22, 6631–6641. [Google Scholar] [CrossRef]
- Kummer, S.; Knoll, A.; Socher, E.; Bethge, L.; Herrmann, A.; Seitz, O. PNA FIT-Probes for the Dual Color Imaging of Two Viral mRNA Targets in Influenza H1N1 Infected Live Cells. Bioconjug. Chem. 2012, 23, 2051–2060. [Google Scholar] [CrossRef]
- Srinivasan, C.; Lee, J.; Papadimitrakopoulos, F.; Silbart, L.K.; Zhao, M.; Burgess, D.J. Labeling and intracellular tracking of functionally active plasmid DNA with semiconductor quantum dots. Mol. Ther. 2006, 14, 192–201. [Google Scholar] [CrossRef]
- Yaroslavsky, A.I.; Smolina, I.V. Fluorescence imaging of single-copy DNA sequences within the human genome using PNA-directed padlock probe assembly. Chem. Biol. 2013, 20, 445–453. [Google Scholar] [CrossRef]
- Meier-Stephenson, V.; Badmalia, M.D.; Mrozowich, T.; Lau, K.C.K.; Schultz, S.K.; Gemmill, D.L.; Osiowy, C.; van Marle, G.; Coffin, C.S.; Patel, T.R. Identification and characterization of a G-quadruplex structure in the pre-core promoter region of hepatitis B virus covalently closed circular DNA. J. Biol. Chem. 2021, 296, 100589. [Google Scholar] [CrossRef]
- Ruggiero, E.; Zanin, I.; Terreri, M.; Richter, S.N. G-Quadruplex Targeting in the Fight against Viruses: An Update. Int. J. Mol. Sci. 2021, 22, 984. [Google Scholar] [CrossRef] [PubMed]
- Yang, D. G-Quadruplex DNA and RNA. Methods Mol. Biol. 2019, 2035, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Xue, B.; Feng, G.; Zhang, J.; Lin, B.; Zeng, P.; Li, H.; Yi, H.; Zhang, X.-L.; Zhu, H.; et al. Lighting up the Native Viral RNA Genome with a Fluorogenic Probe for the Live-Cell Visualization of Virus Infection. J. Am. Chem. Soc. 2019, 141, 5182–5191. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, M.; Zuffo, M.; Nadai, M.; Pirota, V.; Sevilla Montalvo, A.C.; Doria, F.; Freccero, M.; Richter, S.N. Selective targeting of mutually exclusive DNA G-quadruplexes: HIV-1 LTR as paradigmatic model. Nucleic Acids Res. 2020, 48, 4627–4642. [Google Scholar] [CrossRef]
- Huang, G.; Su, C.; Wang, L.; Fei, Y.; Yang, J. The Application of Nucleic Acid Probe–Based Fluorescent Sensing and Imaging in Cancer Diagnosis and Therapy. Front. Chem. 2021, 9, 410. [Google Scholar] [CrossRef]
- Leduc, C.; Si, S.; Gautier, J.J.; Gao, Z.; Shibu, E.S.; Gautreau, A.; Giannone, G.; Cognet, L.; Lounis, B. Chapter 2—Single-molecule imaging in live cell using gold nanoparticles. In Methods in Cell Biology; Paluch, E.K., Ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 125, pp. 13–27. [Google Scholar]
- Ma, Y.; Mao, G.; Huang, W.; Wu, G.; Yin, W.; Ji, X.; Deng, Z.; Cai, Z.; Zhang, X.-E.; He, Z.; et al. Quantum Dot Nanobeacons for Single RNA Labeling and Imaging. J. Am. Chem. Soc. 2019, 141, 13454–13458. [Google Scholar] [CrossRef]
- Zhang, Y.; Ke, X.; Zheng, Z.; Zhang, C.; Zhang, Z.; Zhang, F.; Hu, Q.; He, Z.; Wang, H. Encapsulating Quantum Dots into Enveloped Virus in Living Cells for Tracking Virus Infection. ACS Nano 2013, 7, 3896–3904. [Google Scholar] [CrossRef]
- Chaisomchit, S.; Tyrrell, D.L.; Chang, L.J. Development of replicative and nonreplicative hepatitis B virus vectors. Gene Ther. 1997, 4, 1330–1340. [Google Scholar] [CrossRef]
- Hong, R.; Bai, W.; Zhai, J.; Liu, W.; Li, X.; Zhang, J.; Cui, X.; Zhao, X.; Ye, X.; Deng, Q.; et al. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J. Virol. 2013, 87, 6615–6624. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91 Pt 7, 1854–1864. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleic Acid Form | Description/Encoded | Problems in the Detection | Possibilities to Overcome These Problems |
|---|---|---|---|
| DNA | |||
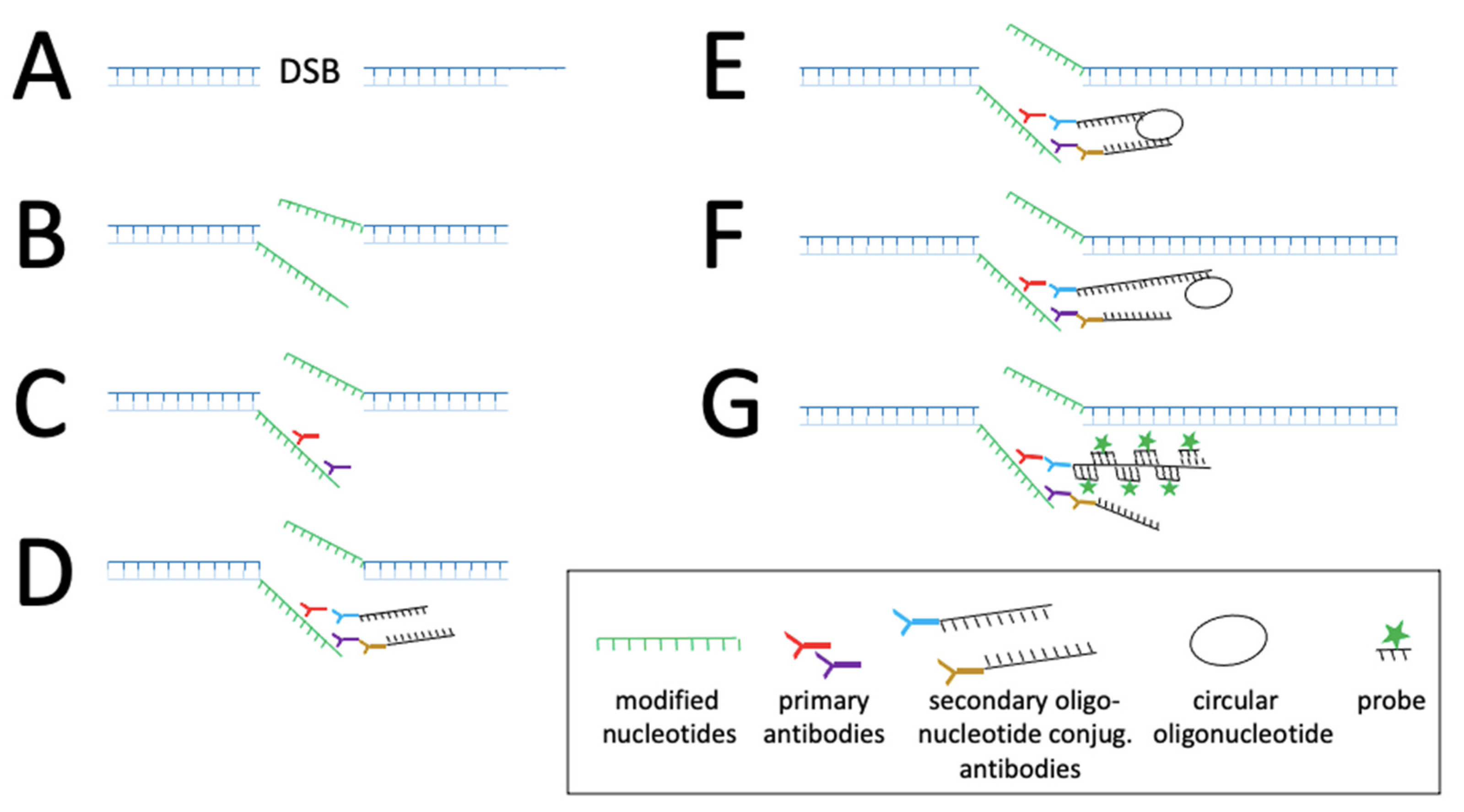
| cccDNA (3.2 kb) | Covalently closed circular DNA Template for progeny viral genomes | (A) Cross reaction with rcDNA, (B) dslDNA, (C) integrated DNA comprises the same sequence than cccDNA but linear. (D) Low copy, high signal/noise ratio | (A) Target the region that is single-stranded in rcDNA. (B, C) Target the region in which the linearization typically occurred. (D) Single molecules detection, amplification of signal, sensors specific of double strand nucleic acids (e.g., CRISPR/STRIDE [23]) |
| rcDNA (3.2 kb) | Relaxed circular DNA | Cross reaction with cccDNA, dslDNA and integrated DNA. | Make assay specific for single strand DNA |
| dslDNA (3.2 kb) | Double-stranded linear DNA | Same sequence as cccDNA but linear | |
| Integrated DNA (3.2 kb or shorter) | Same sequence as cccDNA but linear | ||
| RNA | (A) Low-abundance in contrast with cellular mRNA, high background. (B) Similarity between transcripts: most transcripts from cccDNA use the same poly A site and thereby share the same 3′ end. | (A) Single molecules detection and amplification (e.g., CRISPR/Sunspot system [24]) (B) Discrimination between transcripts can be achieved by combining multiple target regions, e.g., hypothetically by using aptamers (see illustration below), beacons, or designing sgRNAs targeting multiple places in CRISPR system and comparing between the signal from each transcript. | |
| PrecoreRNA (3.5 kb) | Involved in infection and propagation | (A) Cross reaction with other cccDNA derived transcripts. (B) Cross reaction with transcripts from integrated HBV DNA | (A) Combining multiples target (see above) (B) Transcripts from integrated HBV DNA typically start at the start of Pre S, S or X, lack the 3’ end including the typical HBV poly A site and are fusion transcripts using a host gene poly A site. |
| Pre-genomic RNA (3.5 kb) | Template for viral genome Core protein (capsid protein) Polymerase | (A) Cross reaction with other cccDNA derived transcripts (B) Cross reaction with transcripts from integrated HBV DNA. | e.g., |
| Pre S (2.4 kb) | L HBsAg (envelope protein) | (A) Cross reaction with other cccDNA derived transcripts. (B) Cross reaction with transcripts from integrated HBV DNA |  |
| S RNA (2.1 kb) | S HBsAg, M HBsAg (envelope proteins) | (A) Cross reaction with other cccDNA derived transcripts. (B) Cross reaction with transcripts from integrated HBV DNA |  |
| X RNA (0.7 kb) | Regulatory X protein | Cross reaction with other cccDNA derived transcripts (B) Cross reaction with transcripts from integrated HBV DNA. |  |
| From integrations (S) | Cross reaction with cccDNA derived transcripts. |  |
| Technique | Modality | Target | Principal Findings | Advantages | Drawbacks | References |
|---|---|---|---|---|---|---|
| Electron microscopy | cccDNA | • cccDNA exist in heterogeneous population • cccDNA associated with nucleosomes and viral core protein | • High magnification and resolution | • Strong training • Expensive • Low number cccDNA requires enrichment and purification | [29,30] | |
| In situ hybridisation | • Radioactive probes • Biotin • Digoxigenin RNase addition: -without denaturation -with denaturation Co-localisation with HBV antigens • Branched amplification | • Tissue: DNA rcDNA cccDNA and integrated DNA DNA and protein DNA/RNA | • HBV DNA localises mostly in the cytoplasm and with two different patterns • localisation in cytoplasmic • nuclear localisation • Transcription from integrated HBV DNA was shown in HBcAg-negative/ HBsAg-positive cells after hybridization | • Targeting of various genes or biomarkers • Detection of single molecules and two to four target sequences in parallel Higher specificity by using several probe sets | • Deproteinization Mechanical/thermal manipulation • Fixed sample • Low resolution • Differences of sensitivity between antibodies | [31,32,33,34,35,36] [37,38] [39] |
| Fluorescent probes: • FISH -adding capsids -digitonin permeabilized cells adding capsids | • Single cell analysis: DNA/RNA | • HBV genome liberation at nuclear envelope or inside of nucleus • Capsids need nuclear import factors for interacting with nuclear pore complex, and genome release occurs in inner face | • More resolution than autoradiography or enzymatic probes • Can target Targeting of various genes or biomarkers | • No use in living cells. Does not allow kinetic studies of viral genome | [40] [40,41] | |
| • Fluorophore-conjugated readout oligos | • Signal amplification through the binding of two oligos targeting the viral genome in complex with two fluorophores. | [42] | ||||
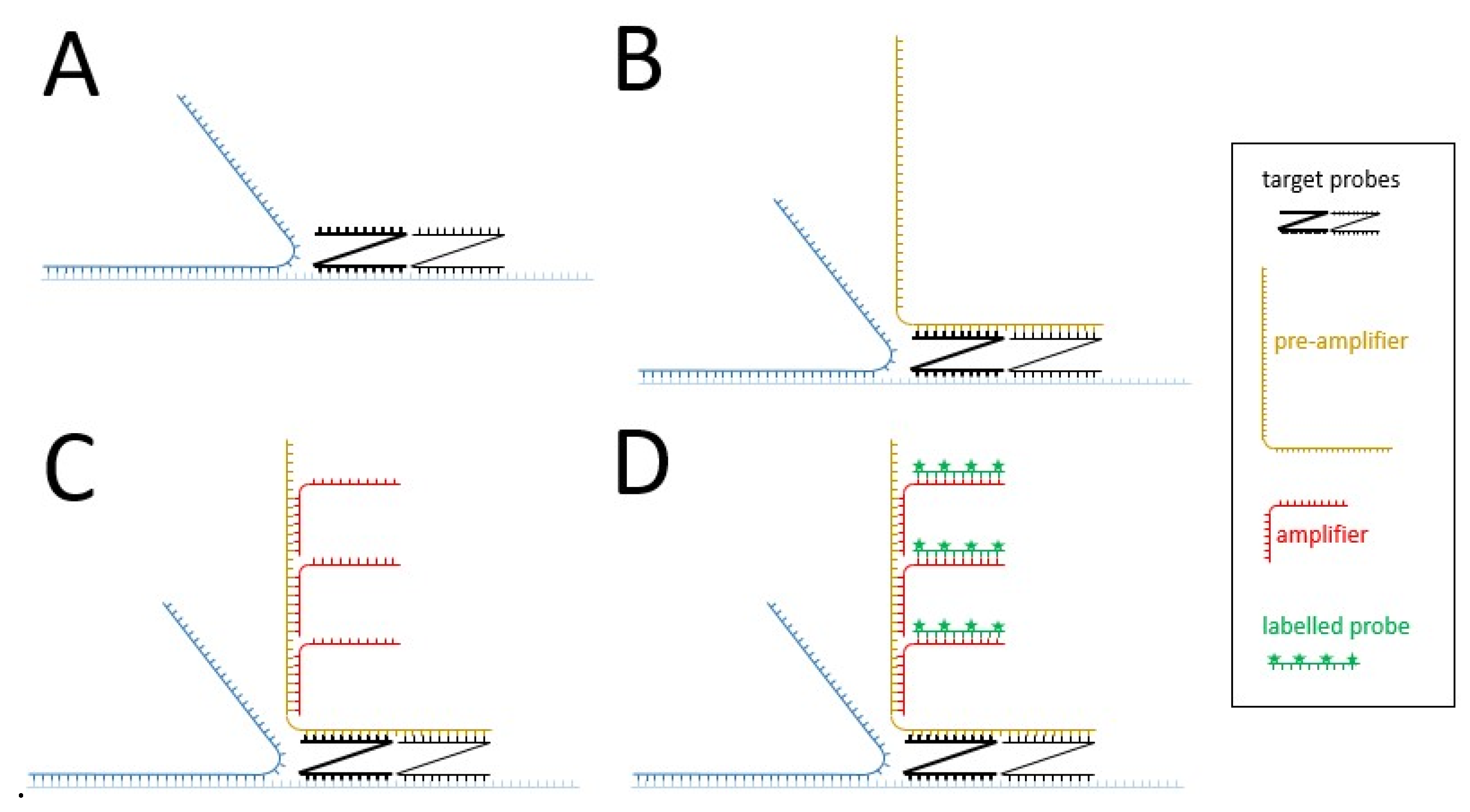
| • bDNA fluorescent | • Distribution and quantification of HBV nucleic acids in the cells | • Signal amplification up to 8000-fold thanks to the branched amplification technique (Figure 2) • More efficient detection than FISH | • Deproteinization | |||
| • RNAscope | • Less time-consuming than ViewRNA ISH | [43,44,45] | ||||
| • ViewRNA ISH | • Time-consuming | [46,47,48,49,50,51] |
| Technique | Modality | Target | Possible Finding | Advantages | Drawbacks | References |
|---|---|---|---|---|---|---|
CRISPR/ Cas | • dCas9-tag: -FP -Sun-tag: -tripartite -Sunspot • sgRNA modification -Aptamers: MS2-MCP, PP7 loops • Cas9 active: -STRIDE | DNA/ RNA dsDNA/ ssDNA | • Single molecule detection • HBV nucleic acids kinetic • Quantification • Distinction between cccDNA and partial DNA | • Fixed and living cells • Allows strategies to improve signal/noise ratio and amplification of signal. • Detection of single molecule and low-abundance of nucleic acids, e.g., Sunspot system • No engineering virus • Specificity • Signal amplification | • Needs signal amplification strategies like Sun-tag system, however large complexes may interfere with nucleic acids kinetic • Requires expression of specific factors in the host cells. • Not applicable in living cells • Antibodies specificity | [52] [53] [23] |
| Non-CRISPR/ Cas | • Aptamer-protein systems | RNA | • Distinction between transcripts (if combined with other methods) | • Fixed and living cells | • Virus engineering • High background • Sensitive folding conditions • Weak photostability | [54] |
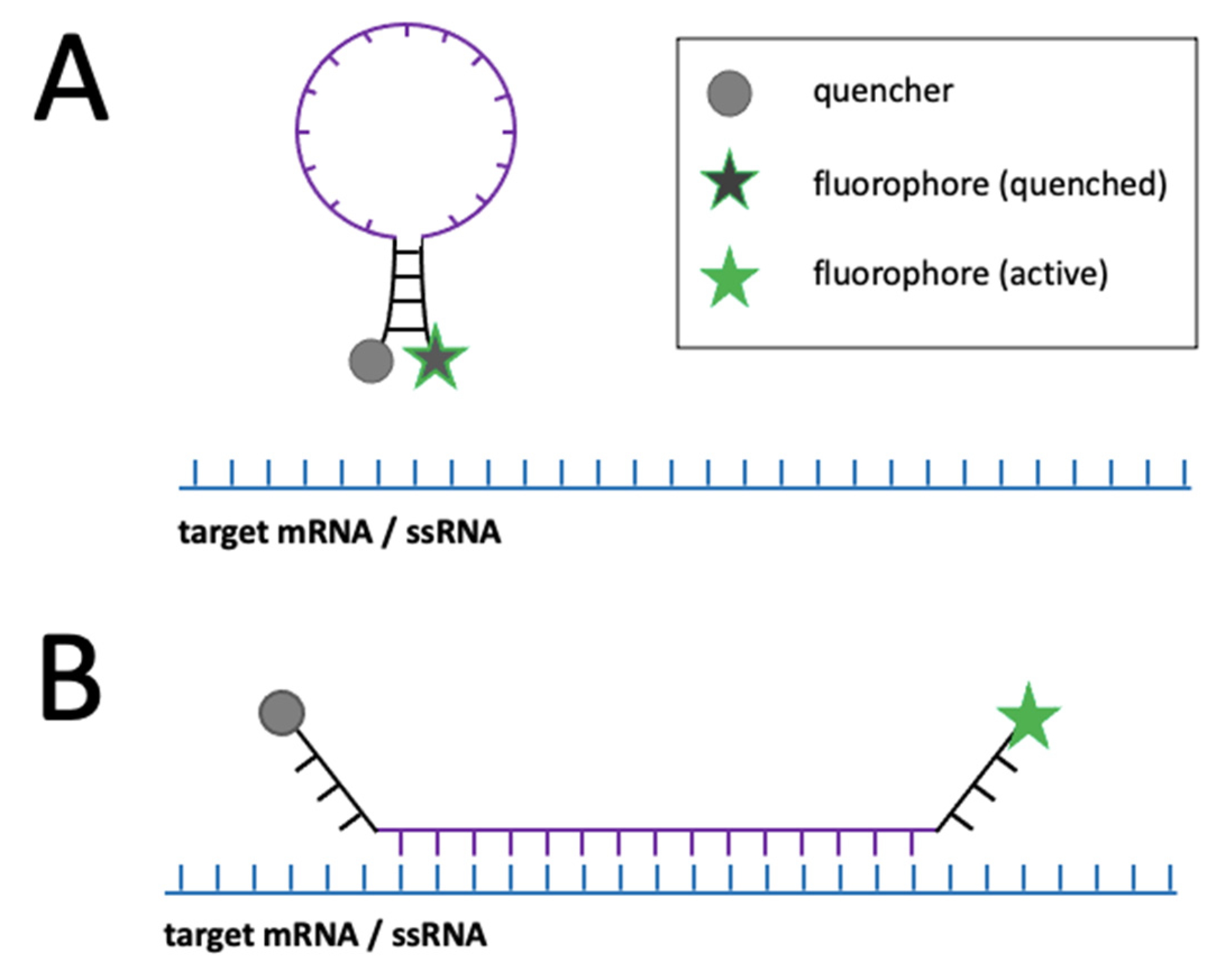
| • Light-up aptamer-dye systems | RNA | • Single-molecules detection possible • Distinction between transcripts (if combined with other methods) | • Fixed and living cells | • Virus engineering • Sensitive folding conditions • Weak photostability | [55,56] | |
| • Molecular beacons | RNA, ssDNA | • Single-molecules detection with advanced microscopes • Distinction between transcripts (if combined with other methods) | • Fixed and living cells | • Prone to nucleases • Difficulties with cell entry • Weak photostability • Prone to non-specific signal | [57,58] | |
| • Quantum-dots / quantum dots-nanobeacons | RNA, ssDNA | • Single-molecule detection • Distinction between transcripts (if combined with other methods) | • Fixed and living cells • Strong signal • Strong photostability | • Cytotoxicity • Difficulties with cell entry due to size • Single-molecules detection | [59,60] | |
| • ANCHOR | dsDNA | • Single-molecule detection • HBV nucleic acids kinetics possible due to photostability • Distinction between cccDNA and rcDNA | • High contrast • Signal amplification • Living cells • Photostability due to bleached molecules replenishment | • Virus engineering • Effect of OR protein on cells unknown • High cytosolic background | ||
| • PNA-based probes | RNA, ssDNA/dsDNA | • Distinction between cccDNA and rcDNA (if combined with other methods) | • Fixed and living cells • High sequence specificity and affinity • Resistant to nucleases • Multiple binding modes make them flexible in function | • Difficulties with cell entry • Prone to non-specific signal to hydrophobic surfaces • Binding mode dependent on careful design | [61] [62] [63] [64] | |
| • Metabolic labeling | dsDNA | • Single-molecules detection • Distinction cccDNA/ rcDNA (if combined with other methods) | • Labelled infectious virus | • Fixed cells only | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante-Jaramillo, L.F.; Fingal, J.; Blondot, M.-L.; Rydell, G.E.; Kann, M. Imaging of Hepatitis B Virus Nucleic Acids: Current Advances and Challenges. Viruses 2022, 14, 557. https://doi.org/10.3390/v14030557
Bustamante-Jaramillo LF, Fingal J, Blondot M-L, Rydell GE, Kann M. Imaging of Hepatitis B Virus Nucleic Acids: Current Advances and Challenges. Viruses. 2022; 14(3):557. https://doi.org/10.3390/v14030557
Chicago/Turabian StyleBustamante-Jaramillo, Luisa F., Joshua Fingal, Marie-Lise Blondot, Gustaf E. Rydell, and Michael Kann. 2022. "Imaging of Hepatitis B Virus Nucleic Acids: Current Advances and Challenges" Viruses 14, no. 3: 557. https://doi.org/10.3390/v14030557
APA StyleBustamante-Jaramillo, L. F., Fingal, J., Blondot, M.-L., Rydell, G. E., & Kann, M. (2022). Imaging of Hepatitis B Virus Nucleic Acids: Current Advances and Challenges. Viruses, 14(3), 557. https://doi.org/10.3390/v14030557