
Low Pathogenicity H7N3 Avian Influenza Viruses Have Higher Within-Host Genetic Diversity Than a Closely Related High Pathogenicity H7N3 Virus in Infected Turkeys and Chickens



Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Samples
2.2. Whole-Genome Sequencing of Avian Influenza Viruses
2.3. Viral Genome Assembly
2.4. Determination of iSNVs
2.5. Calculations for Mean iSNV Frequency and Proportion
2.6. Diversity Measures
2.7. Statistics
3. Results
3.1. Whole Genome Sequencing from Birds Experimentally Infected with H7N3 AIVs
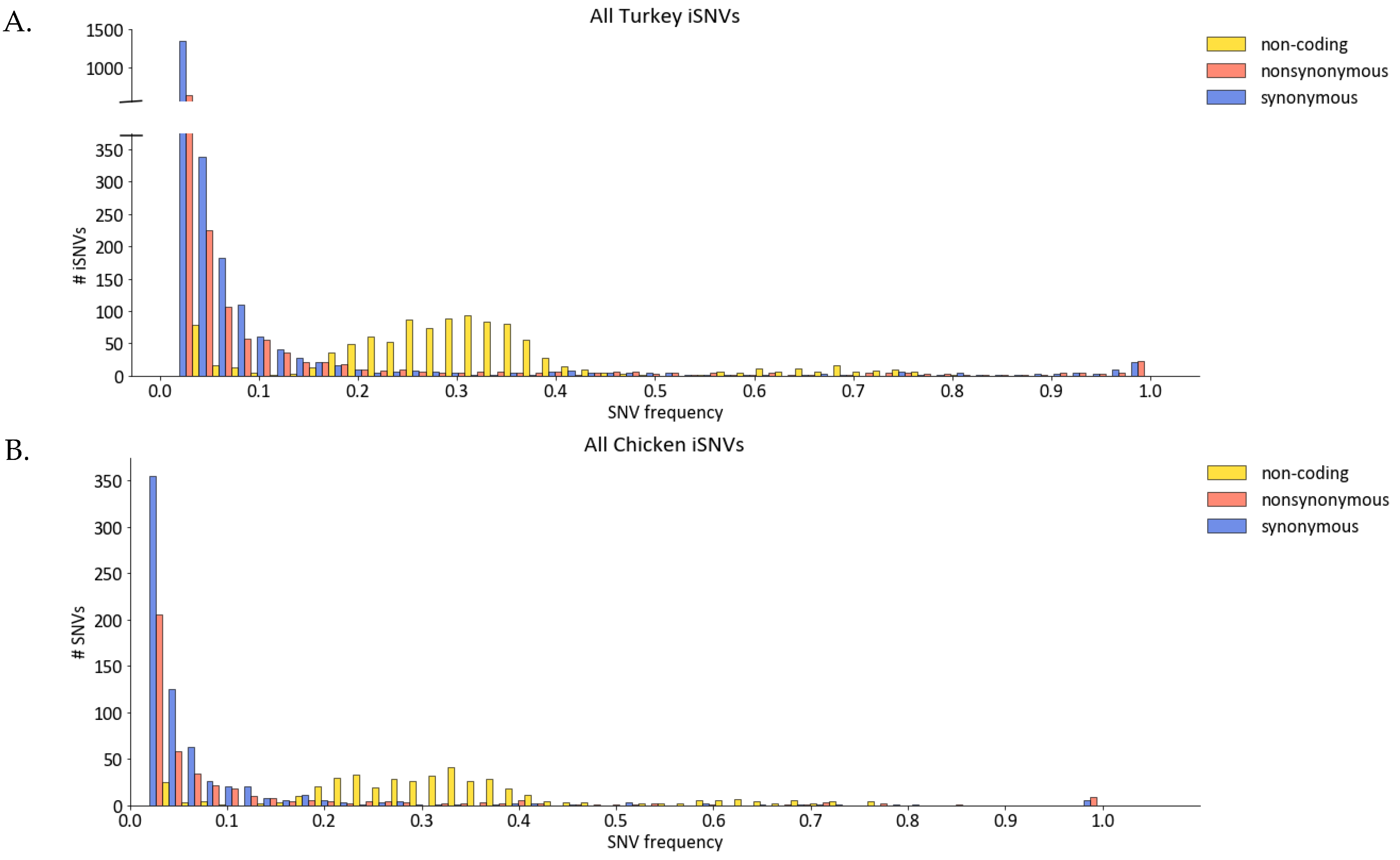
3.2. Most iSNVs Were Nonsynonymous Mutations at Low-Frequency
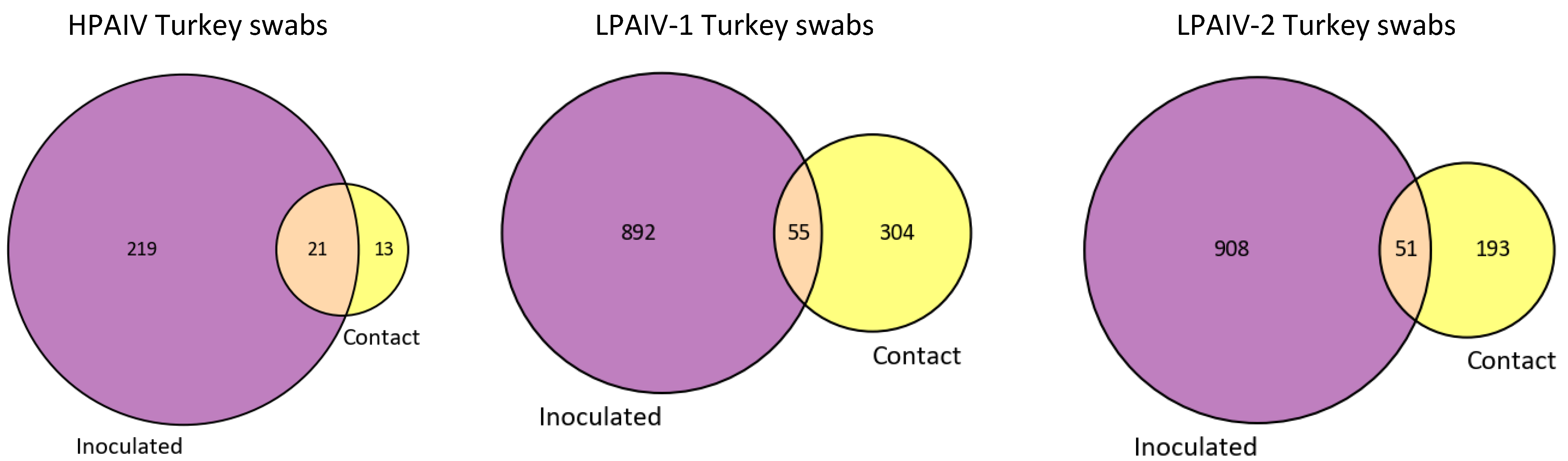
3.3. Only a Few iSNVs Are Shared among Different Host Species and Contact-Exposed Groups
3.4. Comparison of iSNV Diversity with Respect to Host Species, Dose, Route of Exposure, and Time after Infection
3.5. Virus Pathogenicity Type Can Impact Nonsynonymous Changes and iSNV Diversity in Virus Genomes from Directly Inoculated Birds
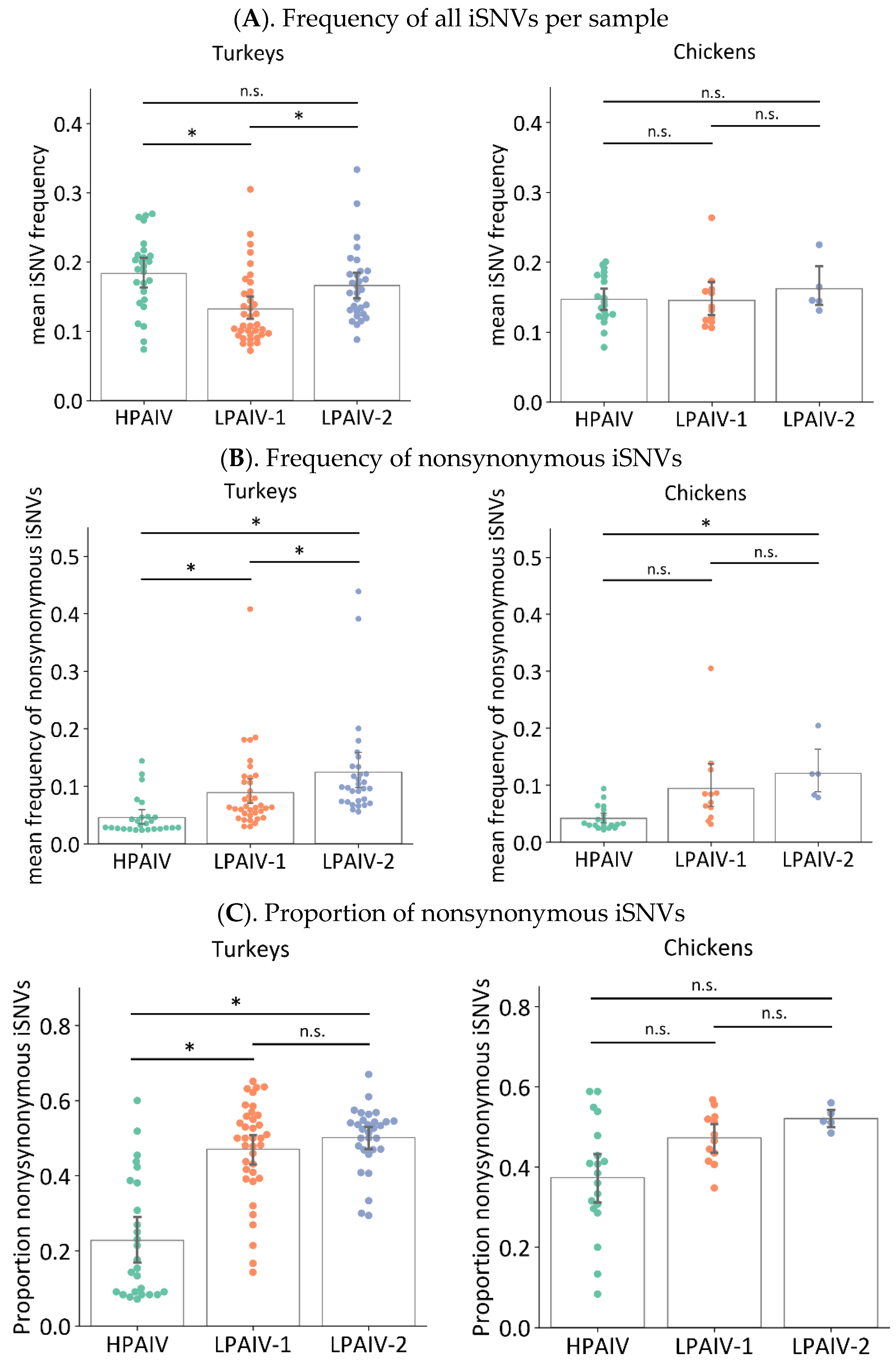
3.5.1. Frequencies and Proportion of Nonsynonymous Changes Are Higher in LPAIVs than HPAIVs in Swabs from Directly Inoculated Birds
3.5.2. The LPAIV Genomes from Swabs Have Higher Genetic Diversity than the HPAIV Genomes
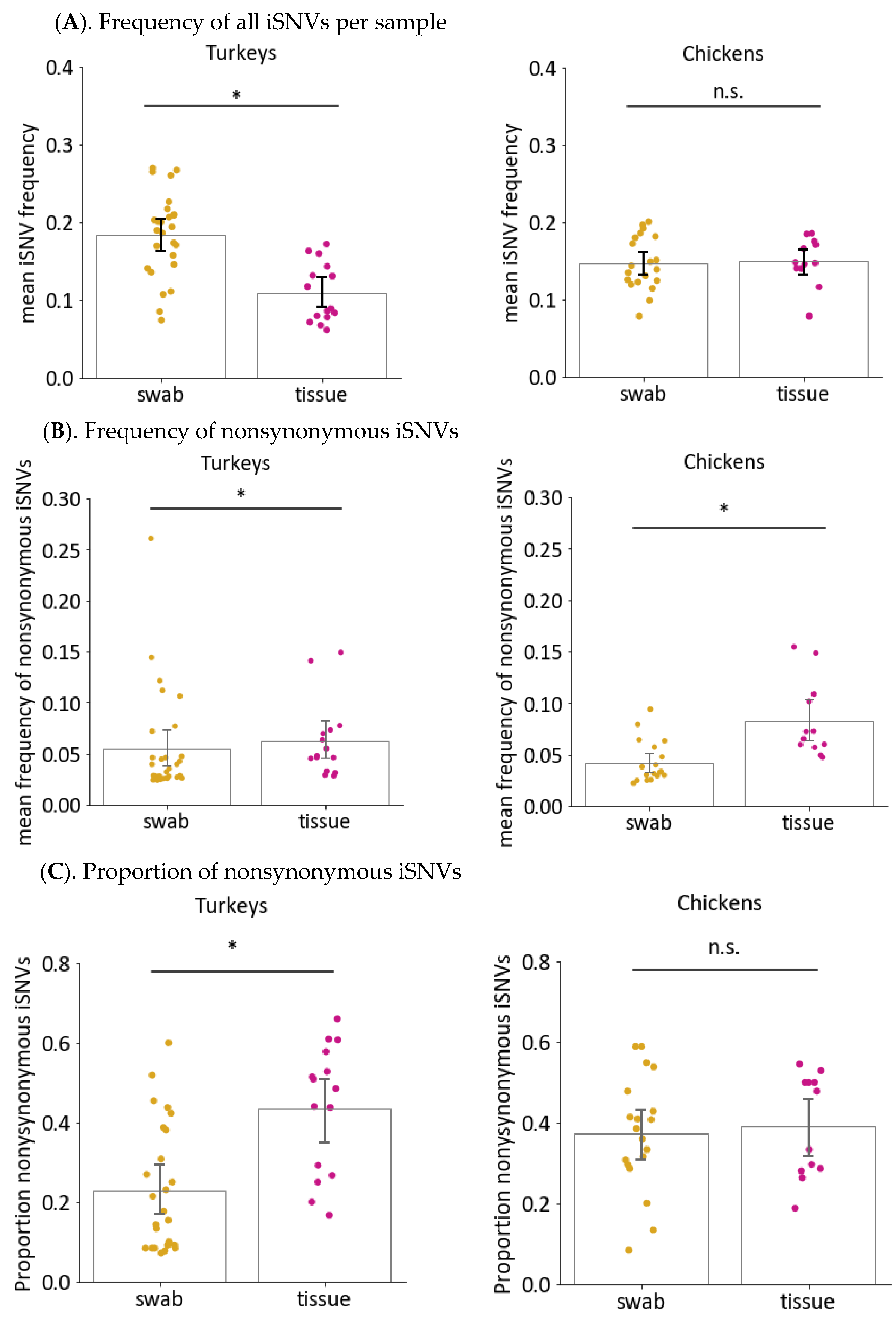
3.6. Sample Type Can also Impact Nonsynonymous Changes and iSNV Diversity in Virus Genomes from Directly Inoculated Birds
3.6.1. Frequencies and Proportion of Nonsynonymous Changes Are Higher in Tissue Samples than Swabs from Turkeys but Not Chickens
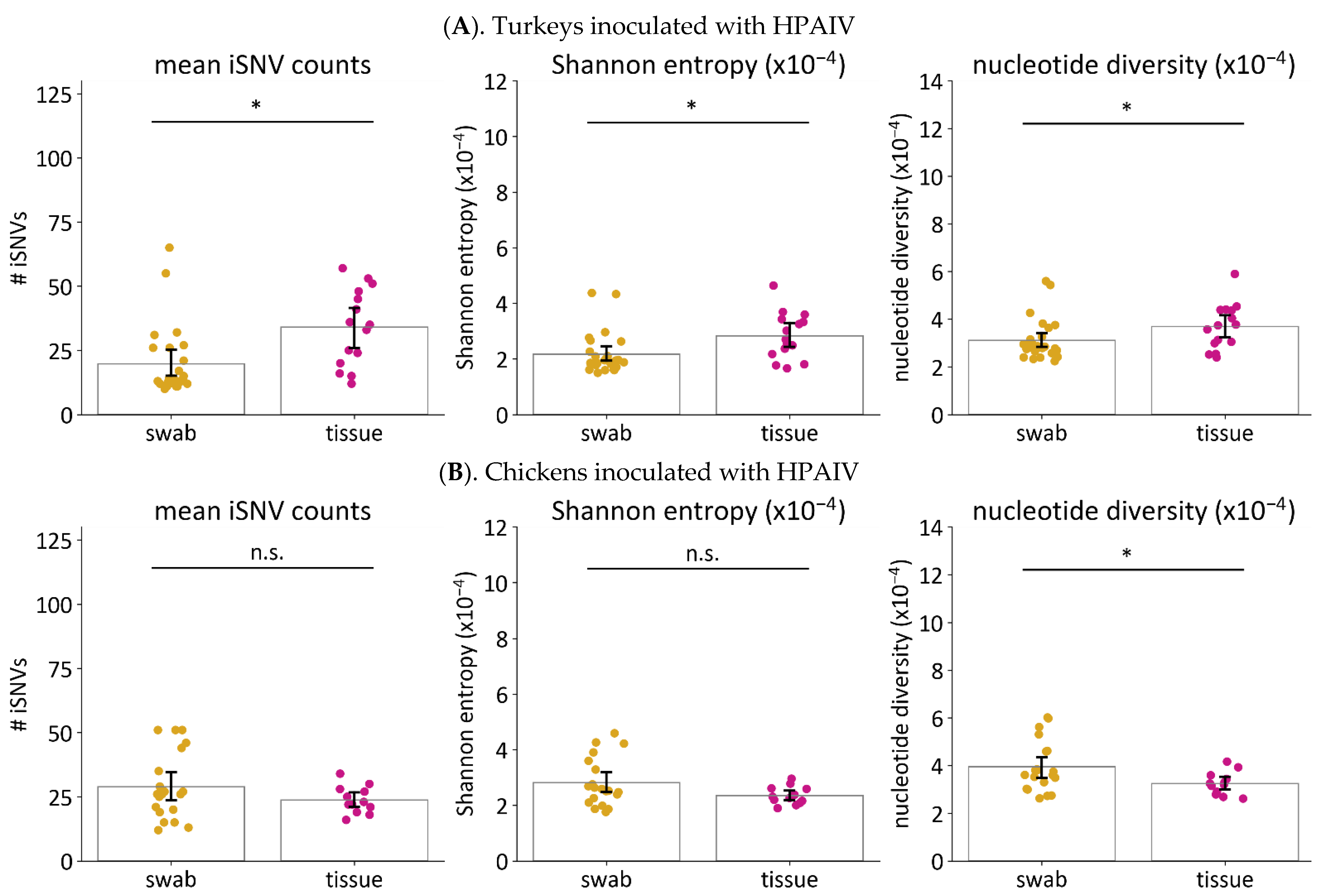
3.6.2. Viruses in Tissues Have Higher Genetic Diversity than Viruses in Swabs from Turkeys Infected with HPAIV
3.7. Some iSNVs in Experimental Samples Were Found in Single Nucleotide Changes in the Outbreak Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Bessière, P.; Volmer, R. From one to many: The within-host rise of viral variants. PLoS Path. 2021, 17, e1009811. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S. Within-Host Viral Diversity: A Window into Viral Evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.W.; Hughes, A.L. Within-host nucleotide diversity of virus populations: Insights from next-generation sequencing. Infect. Genet. Evol. 2015, 30, 1–7. [Google Scholar] [CrossRef]
- Moncla, L.H.; Bedford, T.; Dussart, P.; Horm, S.V.; Rith, S.; Buchy, P.; Karlsson, E.A.; Li, L.; Liu, Y.; Zhu, H.; et al. Quantifying within-host diversity of H5N1 influenza viruses in humans and poultry in Cambodia. PLoS Path. 2020, 16, e1008191. [Google Scholar] [CrossRef]
- Rozek, W.; Kwasnik, M.; Socha, W.; Sztromwasser, P.; Rola, J. Analysis of Single Nucleotide Variants (SNVs) Induced by Passages of Equine Influenza Virus H3N8 in Embryonated Chicken Eggs. Viruses 2021, 13, 1551. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. Elife 2018, 7, e35962. [Google Scholar] [CrossRef]
- Varble, A.; Albrecht, R.A.; Backes, S.; Crumiller, M.; Bouvier, N.M.; Sachs, D.; García-Sastre, A.; Tenoever, B.R. Influenza A Virus Transmission Bottlenecks Are Defined by Infection Route and Recipient Host. Cell Host Microbe 2014, 16, 691–700. [Google Scholar] [CrossRef]
- Leyson, C.; Youk, S.S.; Smith, D.; Dimitrov, K.; Lee, D.H.; Larsen, L.E.; Swayne, D.E.; Pantin-Jackwood, M.J. Pathogenicity and genomic changes of a 2016 European H5N8 highly pathogenic avian influenza virus (clade 2.3.4.4) in experimentally infected mallards and chickens. Virology 2019, 537, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Świętoń, E.; Tarasiuk, K.; Olszewska-Tomczyk, M.; Iwan, E.; Śmietanka, K. A Turkey-origin H9N2 Avian Influenza Virus Shows Low Pathogenicity but Different Within-host Diversity in Experimentally Infected Turkeys, Quail and Ducks. Viruses 2020, 12, 319. [Google Scholar] [CrossRef]
- Valesano, A.L.; Fitzsimmons, W.J.; McCrone, J.T.; Petrie, J.G.; Monto, A.S.; Martin, E.T.; Lauring, A.S. Influenza B Viruses Exhibit Lower Within-Host Diversity than Influenza A Viruses in Human Hosts. J. Virol. 2020, 94, e01710-19. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.; Zhao, L.; Liu, W.; Ding, C.; Nair, V.; Ramos-Onsins, S.E.; Ferretti, L. Genomic Diversity and Evolution of Quasispecies in Newcastle Disease Virus Infections. Viruses 2020, 12, 1305. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Kearney, M.; Palmer, S.; Stephens, R.; Mican, J.; Polis, M.A.; Davey, R.T.; Kovacs, J.; Shao, W.; Rock-Kress, D.; et al. HIV Populations Are Large and Accumulate High Genetic Diversity in a Nonlinear Fashion. J. Virol. 2013, 87, 10313–10323. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Brodin, J.; Thebo, L.; Lanz, C.; Bratt, G.; Albert, J.; Neher, R.A. Population genomics of intrapatient HIV-1 evolution. Elife 2015, 4, e11282. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Puller, V.; Brodin, J.; Albert, J.; Neher, R.A. In vivo mutation rates and the landscape of fitness costs of HIV-1. Virus Evol. 2017, 3, vex003. [Google Scholar] [CrossRef] [PubMed]
- Khera, T.; Todt, D.; Vercauteren, K.; McClure, C.P.; Verhoye, L.; Farhoudi, A.; Bhuju, S.; Geffers, R.; Baumert, T.F.; Steinmann, E.; et al. Tracking HCV protease population diversity during transmission and susceptibility of founder populations to antiviral therapy. Antivir. Res. 2017, 139, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Ueda, Y.; Inuzuka, T.; Yamashita, Y.; Osaki, Y.; Nasu, A.; Umeda, M.; Takemura, R.; Seno, H.; Sekine, A.; et al. Evolution of multi-drug resistant HCV clones from pre-existing resistant-associated variants during direct-acting antiviral therapy determined by third-generation sequencing. Sci. Rep. 2017, 7, 45605. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.J.; Wright, C.F.; Knowles, N.J.; Juleff, N.; Paton, D.J.; King, D.P.; Haydon, D.T. Evolution of foot-and-mouth disease virus intra-sample sequence diversity during serial transmission in bovine hosts. Vet. Res. 2013, 44, 12. [Google Scholar] [CrossRef]
- Braun, K.M.; Moreno, G.K.; Halfmann, P.J.; Hodcroft, E.B.; Baker, D.A.; Boehm, E.C.; Weiler, A.M.; Haj, A.K.; Hatta, M.; Chiba, S.; et al. Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Path. 2021, 17, e1009373. [Google Scholar] [CrossRef] [PubMed]
- Kuny, C.V.; Bowen, C.D.; Renner, D.W.; Johnston, C.M.; Szpara, M.L. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol. 2020, 6, veaa013. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Brese, R.L.; Gonzalez-Perez, M.P.; Koch, M.; O'Connell, O.; Luzuriaga, K.; Somasundaran, M.; Clapham, P.R.; Dollar, J.J.; Nolan, D.J.; Rose, R.; et al. Ultradeep single-molecule real-time sequencing of HIV envelope reveals complete compartmentalization of highly macrophage-tropic R5 proviral variants in brain and CXCR4-using variants in immune and peripheral tissues. J. Neurovirol. 2018, 24, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Brooke, C.B. Biological activities of ‘noninfectious’ influenza A virus particles. Future Virol. 2014, 9, 41–51. [Google Scholar] [CrossRef]
- Swayne, D.E.; Suarez, D.L.; Sims, L.D. Influenza. In Diseases of Poultry; Nair, V., Nolan, L.K., Suarez, D.L., Eds.; John Wiley & Sons, Inc.: Ames, IA, USA, 2013; pp. 181–218. [Google Scholar]
- Avian Influenza. Available online: https://www.oie.int/en/disease/avian-influenza (accessed on 5 January 2021).
- Pasick, J.; Pedersen, J.; Hernandez, M.S. Avian Influenza in North America, 2009–2011. Avian Dis. 2012, 56, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Killian, M.L.; Kim-Torchetti, M.; Hines, N.; Yingst, S.; DeLiberto, T.; Lee, D.H. Outbreak of H7N8 Low Pathogenic Avian Influenza in Commercial Turkeys with Spontaneous Mutation to Highly Pathogenic Avian Influenza. Genome Announc. 2016, 4, e00457-16. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Torchetti, M.K.; Killian, M.L.; Berhane, Y.; Swayne, D.E. Highly Pathogenic Avian Influenza A(H7N9) Virus, Tennessee, USA, March 2017. Emerg. Infect. Dis. 2017, 23, 1860. [Google Scholar] [CrossRef]
- Youk, S.; Lee, D.-H.; Killian, M.; Pantin-Jackwood, M.; Swayne, D.; Torchetti, M. Highly Pathogenic Avian Influenza A(H7N3) Virus in Poultry, United States, 2020. Emerg. Infect. Dis. J. 2020, 26, 2966. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Criado, M.F.; Swayne, D.E. Pathobiological Origins and Evolutionary History of Highly Pathogenic Avian Influenza Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038679. [Google Scholar] [CrossRef]
- Ali, A.; Yassine, H.; Awe, O.O.; Ibrahim, M.; Saif, Y.M.; Lee, C.-W. Replication of swine and human influenza viruses in juvenile and layer turkey hens. Vet. Microbiol. 2013, 163, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Gelb, J., Jr.; Preskenis, L.A.; Ladman, B.S.; Pope, C.R.; Pantin-Jackwood, M.J.; McKinley, E.T. The pathogenesis of low pathogenicity H7 avian influenza viruses in chickens, ducks and turkeys. Virol. J. 2010, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.P.; Pantin-Jackwood, M.; Yassine, H.M.; Saif, Y.M.; Lee, C.W. The high susceptibility of turkeys to influenza viruses of different origins implies their importance as potential intermediate hosts. Avian Dis. 2010, 54, 522–526. [Google Scholar] [CrossRef]
- Criado, M.F.; Leyson, C.M.; Youk, S.; DeBlois, S.; Olivier, T.; Killian, M.L.; Torchetti, M.L.; Parris, D.J.; Spackman, E.; Kapczynski, D.R.; et al. The Pathobiology of H7N3 Low and High Pathogenicity Avian Influenza Viruses from the United States Outbreak in 2020 Differs between Turkeys and Chickens. Viruses 2021, 13, 1851. [Google Scholar] [CrossRef]
- Spackman, E. Avian influenza virus detection and quantitation by real-time RT-PCR. In Animal Influenza Virus: Methods and Protocols; Spackman, E., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 105–118. [Google Scholar]
- Hopken, M.W.; Piaggio, A.J.; Pabilonia, K.L.; Pierce, J.; Anderson, T.; Abdo, Z. Predicting whole genome sequencing success for archived avian influenza virus (Orthomyxoviridae) samples using real-time and droplet PCRs. J. Virol. Methods 2020, 276, 113777. [Google Scholar] [CrossRef]
- Dimitrov, K.M.; Sharma, P.; Volkening, J.D.; Goraichuk, I.V.; Wajid, A.; Rehmani, S.F.; Basharat, A.; Shittu, I.; Joannis, T.M.; Miller, P.J.; et al. A robust and cost-effective approach to sequence and analyze complete genomes of small RNA viruses. Virol. J. 2017, 14, 72. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Lauring, A.S. Measurements of Intrahost Viral Diversity Are Extremely Sensitive to Systematic Errors in Variant Calling. J. Virol. 2016, 90, 6884–6895. [Google Scholar] [CrossRef] [PubMed]
- Sobel Leonard, A.; McClain, M.T.; Smith, G.J.D.; Wentworth, D.E.; Halpin, R.A.; Lin, X.; Ransier, A.; Stockwell, T.B.; Das, S.R.; Gilbert, A.S.; et al. Deep Sequencing of Influenza A Virus from a Human Challenge Study Reveals a Selective Bottleneck and Only Limited Intrahost Genetic Diversification. J. Virol. 2016, 90, 11247–11258. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.; Tassoni, L.; Milani, A.; Hughes, J.; Salviato, A.; Murcia, P.R.; Massi, P.; Zamperin, G.; Bonfanti, L.; Marangon, S.; et al. Unexpected Interfarm Transmission Dynamics during a Highly Pathogenic Avian Influenza Epidemic. J. Virol. 2016, 90, 6401–6411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Illingworth, C.J.R. Measurements of intrahost viral diversity require an unbiased diversity metric. Virus Evol. 2019, 5, vey041. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Koel, B.F.; van Someren Gréve, F.; Vigeveno, R.M.; Pater, M.; Russell, C.A.; de Jong, M.D. Disparate evolution of virus populations in upper and lower airways of mechanically ventilated patients. bioRxiv 2019. bioRxiv:509901. [Google Scholar]
- Xue, K.S.; Moncla, L.H.; Bedford, T.; Bloom, J.D. Within-Host Evolution of Human Influenza Virus. Trends Microbiol. 2018, 26, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021, 13, 30. [Google Scholar] [CrossRef]
- McCune, B.T.; Lanahan, M.R.; tenOever, B.R.; Pfeiffer, J.K. Rapid Dissemination and Monopolization of Viral Populations in Mice Revealed Using a Panel of Barcoded Viruses. J. Virol. 2020, 94, e01590-19. [Google Scholar] [CrossRef]
- Munster, V.J.; Fouchier, R.A.M. Avian influenza virus: Of virus and bird ecology. Vaccine 2009, 27, 6340–6344. [Google Scholar] [CrossRef]
- World Health Organisation. Avian influenza (Infection with avian influenza viruses). In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Swayne, D.E.; Suarez, D.L. Highly pathogenic avian influenza. Int. Off. Epizoot. 2000, 19, 463–482. [Google Scholar] [CrossRef]
- Dlugolenski, D.; Jones, L.; Saavedra, G.; Tompkins, S.M.; Tripp, R.A.; Mundt, E. Passage of low-pathogenic avian influenza (LPAI) viruses mediates rapid genetic adaptation of a wild-bird isolate in poultry. Arch. Virol. 2011, 156, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Renner, D.W.; Szpara, M.L.; Glaunsinger, B.A. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92, e00908-17. [Google Scholar] [CrossRef] [PubMed]
- Sobel Leonard, A.; Weissman, D.B.; Greenbaum, B.; Ghedin, E.; Koelle, K. Transmission Bottleneck Size Estimation from Pathogen Deep-Sequencing Data, with an Application to Human Influenza A Virus. J. Virol. 2017, 91, e00171-17. [Google Scholar] [CrossRef] [PubMed]
- Wilker, P.R.; Dinis, J.M.; Starrett, G.; Imai, M.; Hatta, M.; Nelson, C.W.; O'Connor, D.H.; Hughes, A.L.; Neumann, G.; Kawaoka, Y.; et al. Selection on haemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat. Commun. 2013, 4, 2636. [Google Scholar] [CrossRef][Green Version]
- Dinis, J.M.; Florek, N.W.; Fatola, O.O.; Moncla, L.H.; Mutschler, J.P.; Charlier, O.K.; Meece, J.K.; Belongia, E.A.; Friedrich, T.C. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J. Virol. 2016, 90, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Holmes, E.C. Avian Influenza Virus Exhibits Rapid Evolutionary Dynamics. Mol. Biol. Evol. 2006, 23, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- Suarez, D.L. Evolution of avian influenza viruses. Vet. Microbiol. 2000, 74, 15–27. [Google Scholar] [CrossRef]
- Pauly, M.D.; Procario, M.C.; Lauring, A.S. A novel twelve class fluctuation test reveals higher than expected mutation rates for influenza A viruses. eLife 2017, 6, e26437. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.-S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (Favipiravir) Induces Lethal Mutagenesis in Influenza A H1N1 Viruses In Vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef]
- Cheung, P.P.H.; Watson, S.J.; Choy, K.-T.; Fun Sia, S.; Wong, D.D.Y.; Poon, L.L.M.; Kellam, P.; Guan, Y.; Malik Peiris, J.S.; Yen, H.-L. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat. Commun. 2014, 5, 4794. [Google Scholar] [CrossRef]
- Pauly, M.D.; Lauring, A.S.; Dermody, T.S. Effective Lethal Mutagenesis of Influenza Virus by Three Nucleoside Analogs. J. Virol. 2015, 89, 3584–3597. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jiang, D.; Ni, M.; Li, J.; Chen, Z.; Liu, J.; Ye, H.; Wong, G.; Li, W.; Zhang, Y.; et al. Phylogenomic analysis unravels evolution of yellow fever virus within hosts. PLoS Negl. Trop. Dis. 2018, 12, e0006738. [Google Scholar] [CrossRef] [PubMed]
- Sall, A.A.; Faye, O.; Diallo, M.; Firth, C.; Kitchen, A.; Holmes, E.C. Yellow Fever Virus Exhibits Slower Evolutionary Dynamics than Dengue Virus. J. Virol. 2010, 84, 765–772. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.; Block, A.; Beaucourt, S.; Moratorio, G.; Vignuzzi, M.; Peersen, O.B. Design of a Genetically Stable High Fidelity Coxsackievirus B3 Polymerase That Attenuates Virus Growth In Vivo. J. Biol. Chem. 2016, 291, 13999–14011. [Google Scholar] [CrossRef]
- Tsai, Y.-H.; Huang, S.-W.; Hsieh, W.-S.; Cheng, C.-K.; Chang, C.-F.; Wang, Y.-F.; Wang, J.-R.; Pfeiffer, J.K. Enterovirus A71 Containing Codon-Deoptimized VP1 and High-Fidelity Polymerase as Next-Generation Vaccine Candidate. J. Virol. 2019, 93, e02308-18. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased Fidelity Reduces Poliovirus Fitness and Virulence under Selective Pressure in Mice. PLoS Path. 2005, 1, e11. [Google Scholar] [CrossRef]
- Fourment, M.; Holmes, E.C. Avian influenza virus exhibits distinct evolutionary dynamics in wild birds and poultry. BMC Evol. Biol. 2015, 15, 120. [Google Scholar] [CrossRef]
- Youk, S.; Lee, D.H.; Ferreira, H.L.; Afonso, C.L.; Absalon, A.E.; Swayne, D.E.; Suarez, D.L.; Pantin-Jackwood, M.J. Rapid evolution of Mexican H7N3 highly pathogenic avian influenza viruses in poultry. PLoS ONE 2019, 14, e0222457. [Google Scholar] [CrossRef]
- Barnard, K.N.; Wasik, B.R.; Alford-Lawrence, B.K.; Hayward, J.J.; Weichert, W.S.; Voorhees, I.E.H.; Holmes, E.C.; Parrish, C.R. Sequence dynamics of three influenza A virus strains grown in different MDCK cell lines, including those expressing different sialic acid receptors. J. Evol. Biol. 2021, 34, 1878–1900. [Google Scholar] [CrossRef]
- Hopken, M.W.; Piaggio, A.J.; Pabilonia, K.L.; Pierce, J.; Anderson, T.; Pierce, C.; Abdo, Z. Population genomic transformations induced by isolation of wild bird avian influenza viruses (Orthomyxoviridae) in embryonated chicken eggs. Infect. Genet. Evol. 2020, 90, 104505. [Google Scholar] [CrossRef]
- Russell, G.C.; Zadoks, R.N.; Willoughby, K.; Bachofen, C. Bovine viral diarrhoea virus loses quasispecies diversity rapidly in culture. Microbial Genom. 2020, 6, e000343. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Abel zur Wiesch, P.; Davis, B.M.; Waldor, M.K. Analysis of Bottlenecks in Experimental Models of Infection. PLoS Path. 2015, 11, e1004823. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Lauring, A.S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 2018, 28, 20–25. [Google Scholar] [CrossRef]
- da Silva, W.; Kutnjak, D.; Xu, Y.; Xu, Y.; Giovannoni, J.; Elena, S.F.; Gray, S. Transmission modes affect the population structure of potato virus Y in potato. PLoS Path. 2020, 16, e1008608. [Google Scholar] [CrossRef] [PubMed]
- Aimone, C.D.; Lavington, E.; Hoyer, J.S.; Deppong, D.O.; Mickelson-Young, L.; Jacobson, A.; Kennedy, G.G.; Carbone, I.; Hanley-Bowdoin, L.; Duffy, S. Population diversity of cassava mosaic begomoviruses increases over the course of serial vegetative propagation. J. Gen. Virol. 2021, 102, 001622. [Google Scholar] [CrossRef] [PubMed]
- Gelbart, M.; Harari, S.; Ben-Ari, Y.; Kustin, T.; Wolf, D.; Mandelboim, M.; Mor, O.; Pennings, P.S.; Stern, A. Drivers of within-host genetic diversity in acute infections of viruses. PLoS Path. 2020, 16, e1009029. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, H.A.; Benslimane, F.M.; Elbashir, I.E.; Coyle, P.V.; Al Maslamani, M.A.; Al-Khal, A.; Al Thani, A.A.; Yassine, H.M. Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients With Variable Disease Severities. Front. Cell. Infect. Microbiol. 2020, 10, 534. [Google Scholar] [CrossRef]
- Banks, J.; Speidel, E.S.; Moore, E.; Plowright, L.; Piccirillo, A.; Capua, I.; Cordioli, P.; Fioretti, A.; Alexander, D.J. Changes in the haemagglutinin and the neuraminidase genes prior to the emergence of highly pathogenic H7N1 avian influenza viruses in Italy. Arch. Virol. 2001, 146, 963–973. [Google Scholar] [CrossRef]
- Campitelli, L.; Mogavero, E.; De Marco, M.A.; Delogu, M.; Puzelli, S.; Frezza, F.; Facchini, M.; Chiapponi, C.; Foni, E.; Cordioli, P.; et al. Interspecies transmission of an H7N3 influenza virus from wild birds to intensively reared domestic poultry in Italy. Virology 2004, 323, 24–36. [Google Scholar] [CrossRef]
- Giannecchini, S.; Clausi, V.; Di Trani, L.; Falcone, E.; Terregino, C.; Toffan, A.; Cilloni, F.; Matrosovich, M.; Gambaryan, A.S.; Bovin, N.V.; et al. Molecular adaptation of an H7N3 wild duck influenza virus following experimental multiple passages in quail and turkey. Virology 2010, 408, 167–173. [Google Scholar] [CrossRef]
- Li, J.; Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and Genetic Variation of Neuraminidase Stalk Deletions in Avian Influenza Viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.W.; Munier, S.; Larcher, T.; Soubieux, D.; Ledevin, M.; Esnault, E.; Tourdes, A.; Croville, G.; Guérin, J.-L.; Quéré, P.; et al. Length Variations in the NA Stalk of an H7N1 Influenza Virus Have Opposite Effects on Viral Excretion in Chickens and Ducks. J. Virol. 2012, 86, 584–588. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Species | Exposure | Sample Type | # Samples a | # Birds b |
|---|---|---|---|---|---|
| LPAIV-1 | Turkey | inoculated | OP | 37/56 | 10/10 |
| Chicken | OP | 12/33 | 6/9 | ||
| LPAIV-2 | Turkey | OP | 30/94 | 13/15 | |
| Chicken | OP | 5/30 | 5/10 | ||
| HPAIV | Turkey | OP | 26/33 | 15/15 | |
| tissues | 15/15 | 3/3 | |||
| Chicken | OP | 20/23 | 13/14 | ||
| tissues | 12/15 | 3/3 | |||
| LPAIV-1 | Turkey | contact | OP | 13/17 | 2/2 |
| LPAIV-2 | OP | 6/17 | 2/2 | ||
| HPAIV | OP | 4/12 | 2/2 | ||
| Total | 180/345 | 74/85 |
| Number of Samples a | ||||||||
|---|---|---|---|---|---|---|---|---|
| Turkeys | Chickens | |||||||
| Inoculated | Contact | Inoculated | ||||||
| Virus | Segment | SNV | Amino Acid Change | OP | Tissue | OP | OP | Tissue |
| LPAIV-1 | NP | T507C | synonymous | 2/37 | n.a. | 1/13 | 3/12 | n.a. |
| LPAIV-2 | PB1 | G183A | PB1-F2 G22E | 4/30 | n.a. | 0/6 | 1/5 | n.a. |
| LPAIV-2 | NS | G485A | E153K | 19/30 | n.a. | 6/6 | 2/5 | n.a. |
| HPAIV | PA | T1489A | C489S | 0/26 | 3/15 | 0/4 | 0/20 | 2/12 |
| HPAIV | NS | T294C | L90P | 6/26 | 5/15 | 2/4 | 3/20 | 0/12 |
| Sample Type | Virus | Species | iSNV Frequency | Nonsynonymous iSNV Frequency | Proportion of Nonsynonymous iSNVs | SNV Count | Shannon Entropy (×10−4) | Nucleotide Diversity (×10−4) |
|---|---|---|---|---|---|---|---|---|
| swab | HPAIV | Chicken | 0.15 | 0.045 | 0.374 | 28.95 | 2.81 | 3.95 |
| Turkey | 0.19 | 0.050 | 0.231 | 19.23 | 2.16 | 3.10 | ||
| LPAIV-1 | Chicken | 0.15 | 0.084 | 0.473 | 38.00 | 3.90 | 5.41 | |
| Turkey | 0.14 | 0.080 | 0.477 | 37.86 | 3.28 | 4.40 | ||
| LPAIV-2 | Chicken | 0.16 | 0.116 | 0.521 | 38.60 | 3.89 | 5.43 | |
| Turkey | 0.16 | 0.099 | 0.511 | 47.53 | 4.29 | 5.83 | ||
| tissue | HPAIV | Chicken | 0.15 | 0.085 | 0.392 | 23.75 | 2.35 | 3.25 |
| Turkey | 0.11 | 0.056 | 0.436 | 34.07 | 2.83 | 3.69 | ||
| inocula | HPAIV | n.a. | 0.11 | 0.065 | 0.281 | 48.00 | 4.42 | 5.93 |
| LPAIV-1 | n.a. | 0.11 | 0.071 | 0.255 | 41.00 | 3.59 | 4.74 | |
| LPAIV-2 | n.a. | 0.10 | 0.080 | 0.307 | 50.00 | 4.26 | 5.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leyson, C.M.; Criado, M.F.; Youk, S.; Pantin-Jackwood, M.J. Low Pathogenicity H7N3 Avian Influenza Viruses Have Higher Within-Host Genetic Diversity Than a Closely Related High Pathogenicity H7N3 Virus in Infected Turkeys and Chickens. Viruses 2022, 14, 554. https://doi.org/10.3390/v14030554
Leyson CM, Criado MF, Youk S, Pantin-Jackwood MJ. Low Pathogenicity H7N3 Avian Influenza Viruses Have Higher Within-Host Genetic Diversity Than a Closely Related High Pathogenicity H7N3 Virus in Infected Turkeys and Chickens. Viruses. 2022; 14(3):554. https://doi.org/10.3390/v14030554
Chicago/Turabian StyleLeyson, Christina M., Miriã F. Criado, Sungsu Youk, and Mary J. Pantin-Jackwood. 2022. "Low Pathogenicity H7N3 Avian Influenza Viruses Have Higher Within-Host Genetic Diversity Than a Closely Related High Pathogenicity H7N3 Virus in Infected Turkeys and Chickens" Viruses 14, no. 3: 554. https://doi.org/10.3390/v14030554
APA StyleLeyson, C. M., Criado, M. F., Youk, S., & Pantin-Jackwood, M. J. (2022). Low Pathogenicity H7N3 Avian Influenza Viruses Have Higher Within-Host Genetic Diversity Than a Closely Related High Pathogenicity H7N3 Virus in Infected Turkeys and Chickens. Viruses, 14(3), 554. https://doi.org/10.3390/v14030554