
Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression

 , ,
, ,  , , , ,
, , , ,  and add
Show full author list
and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
3. Results
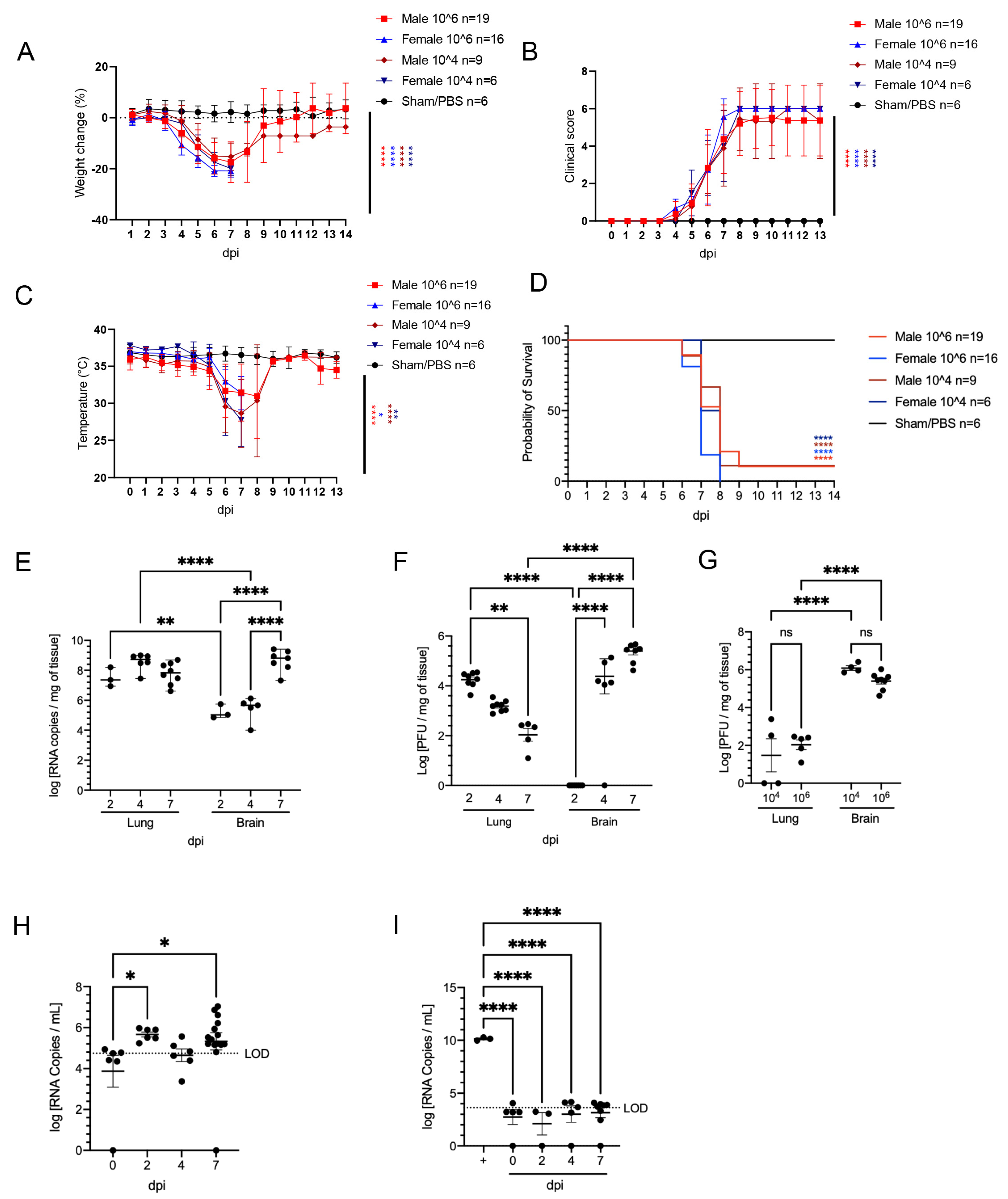
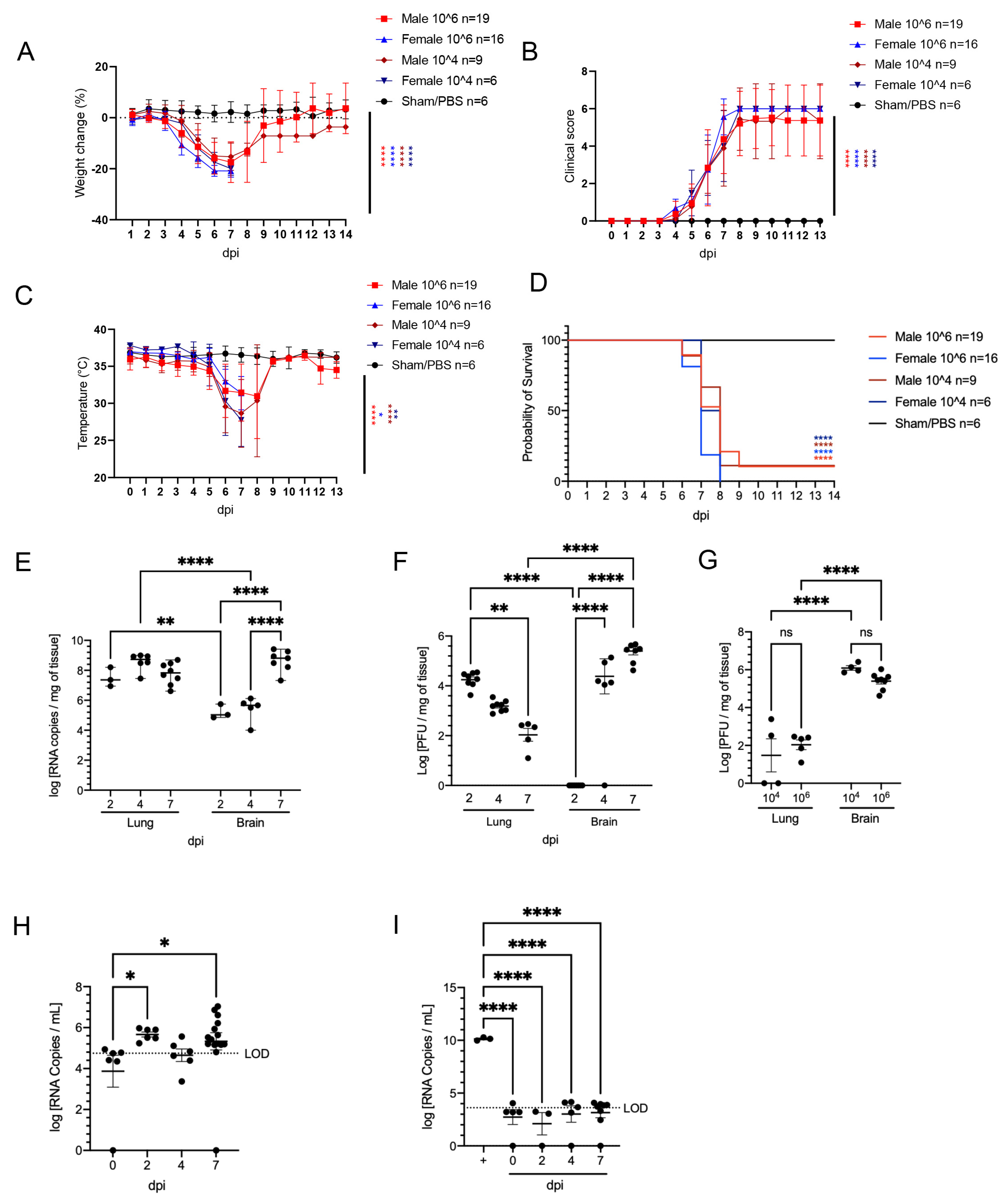
3.1. SARS-CoV-2 Is Nearly Invariably Fatal in Infected K18-hACE2 Mice over a 104–106 PFU Inoculation Dose
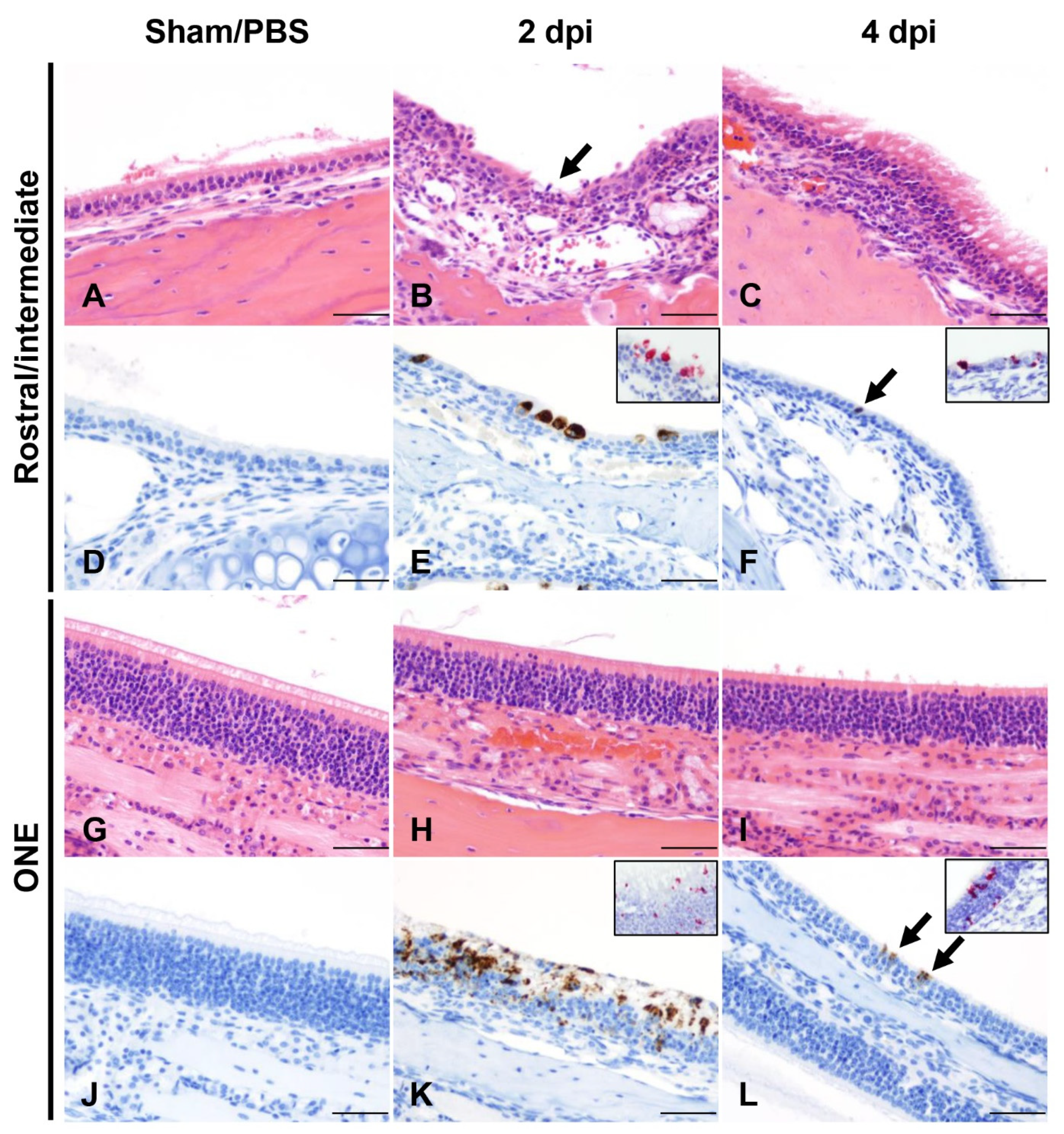
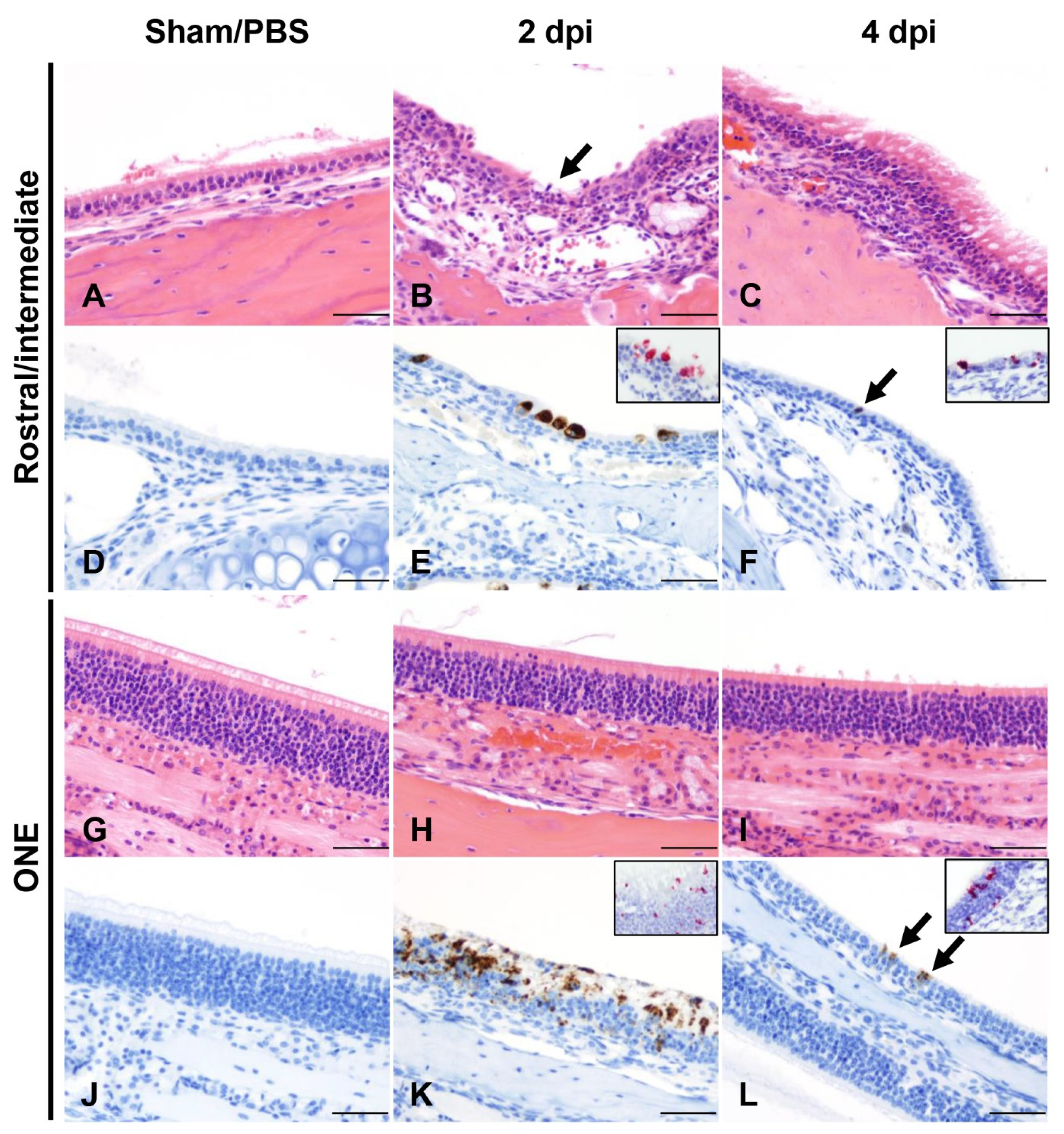
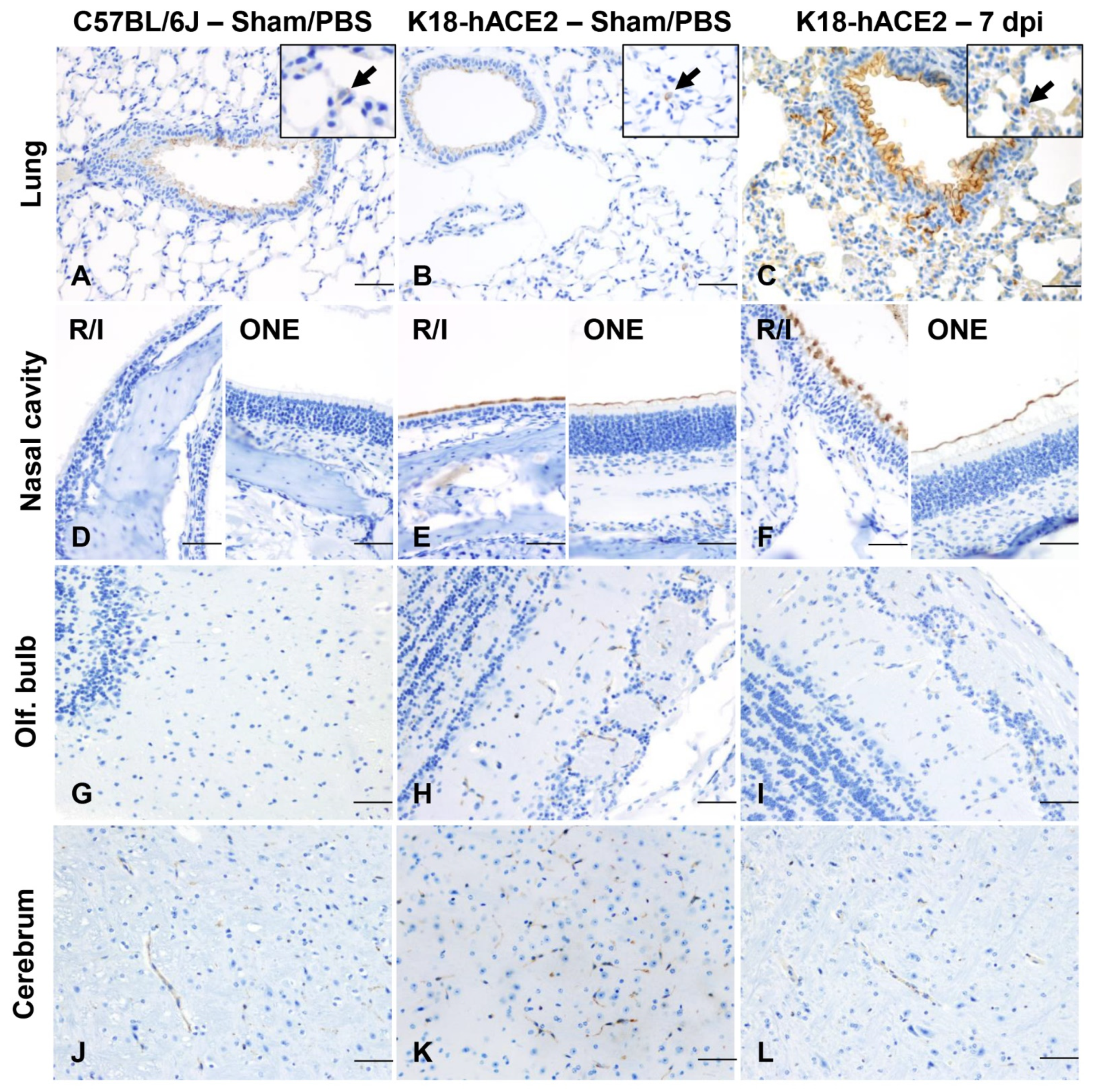
3.2. SARS-CoV-2 Results in Transient Mild Infection in the Nasal Cavity of K18-hACE2 Mice
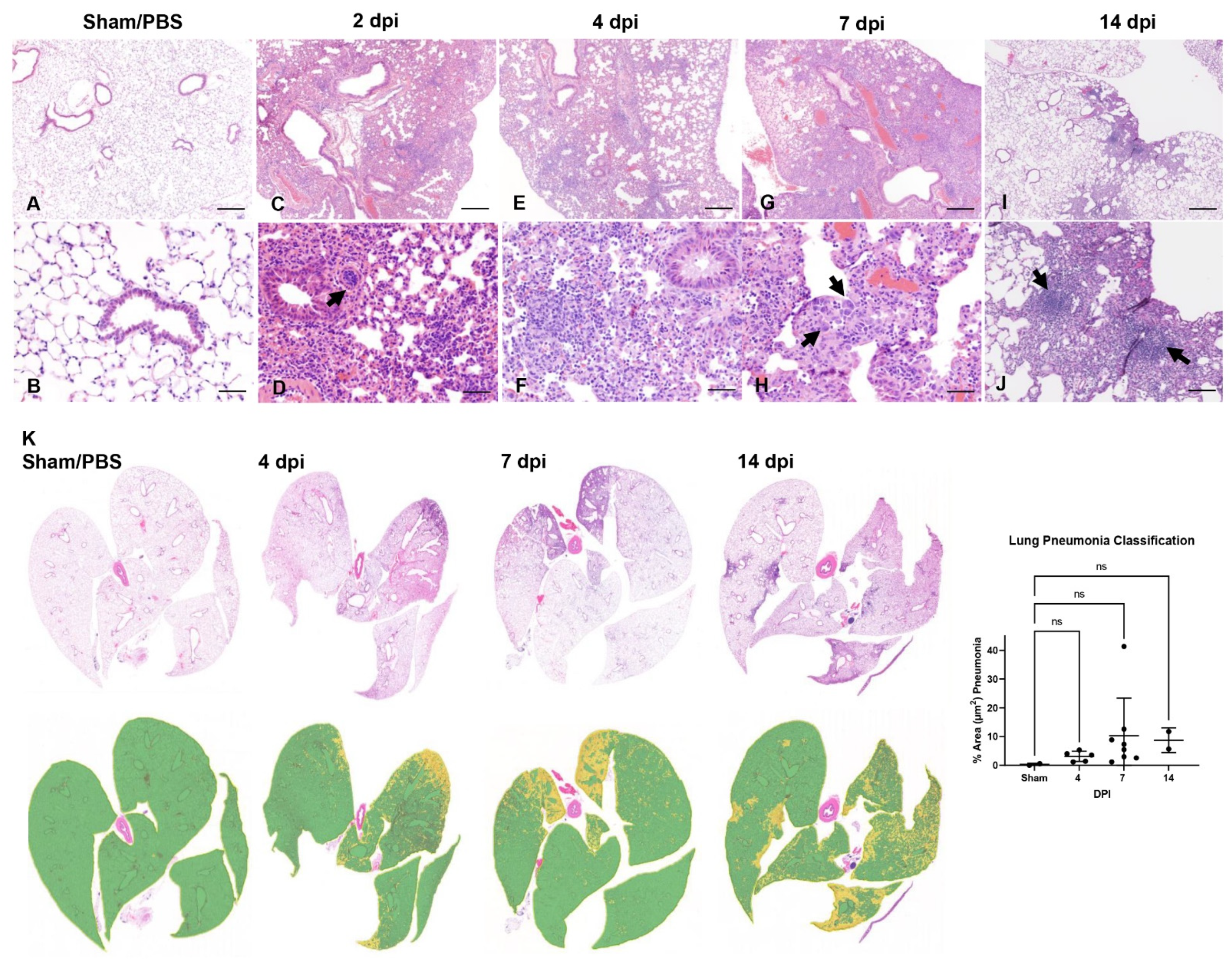
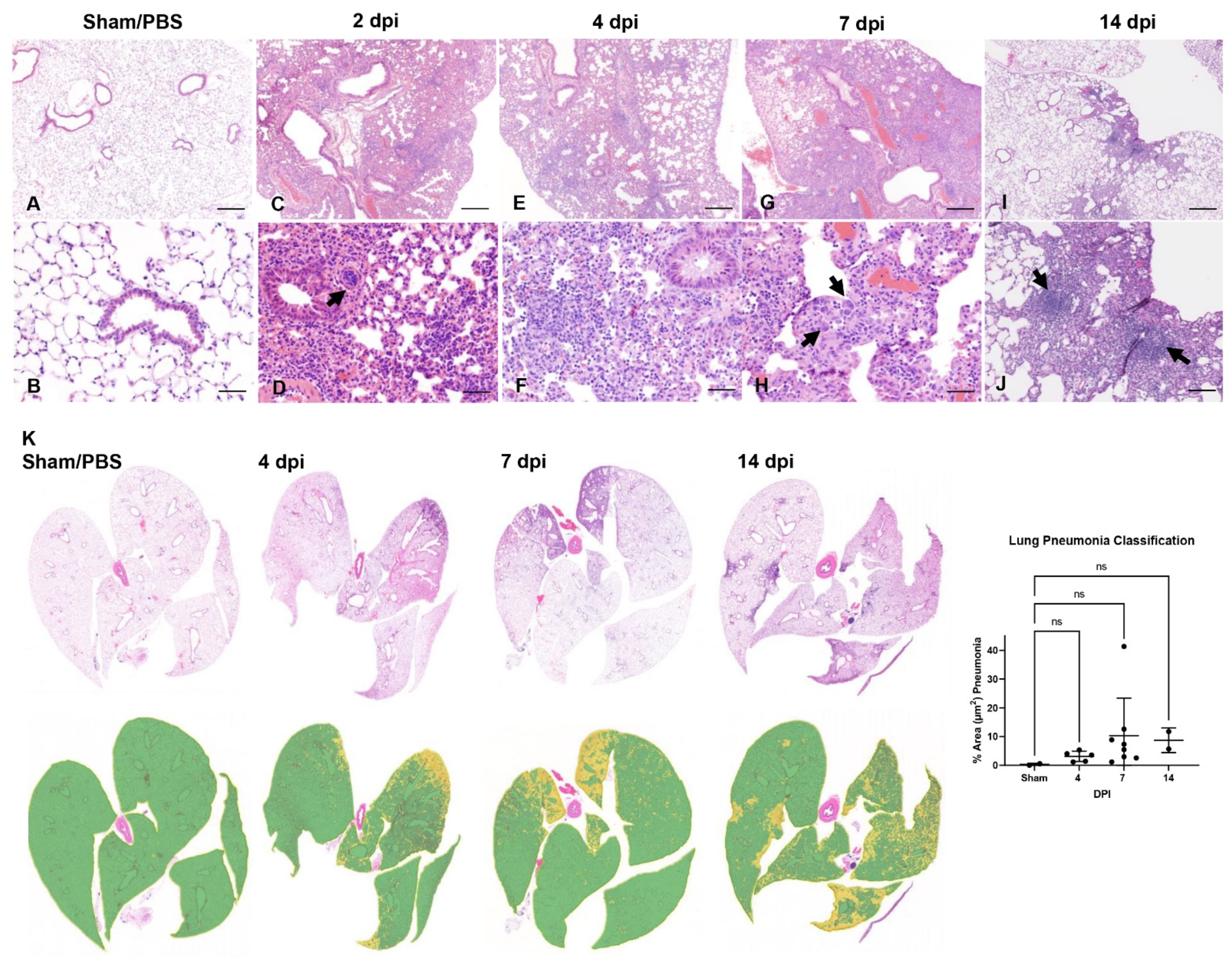
3.3. SARS-CoV-2 Induces Moderate Interstitial Pneumonia in K18-hACE2 Mice
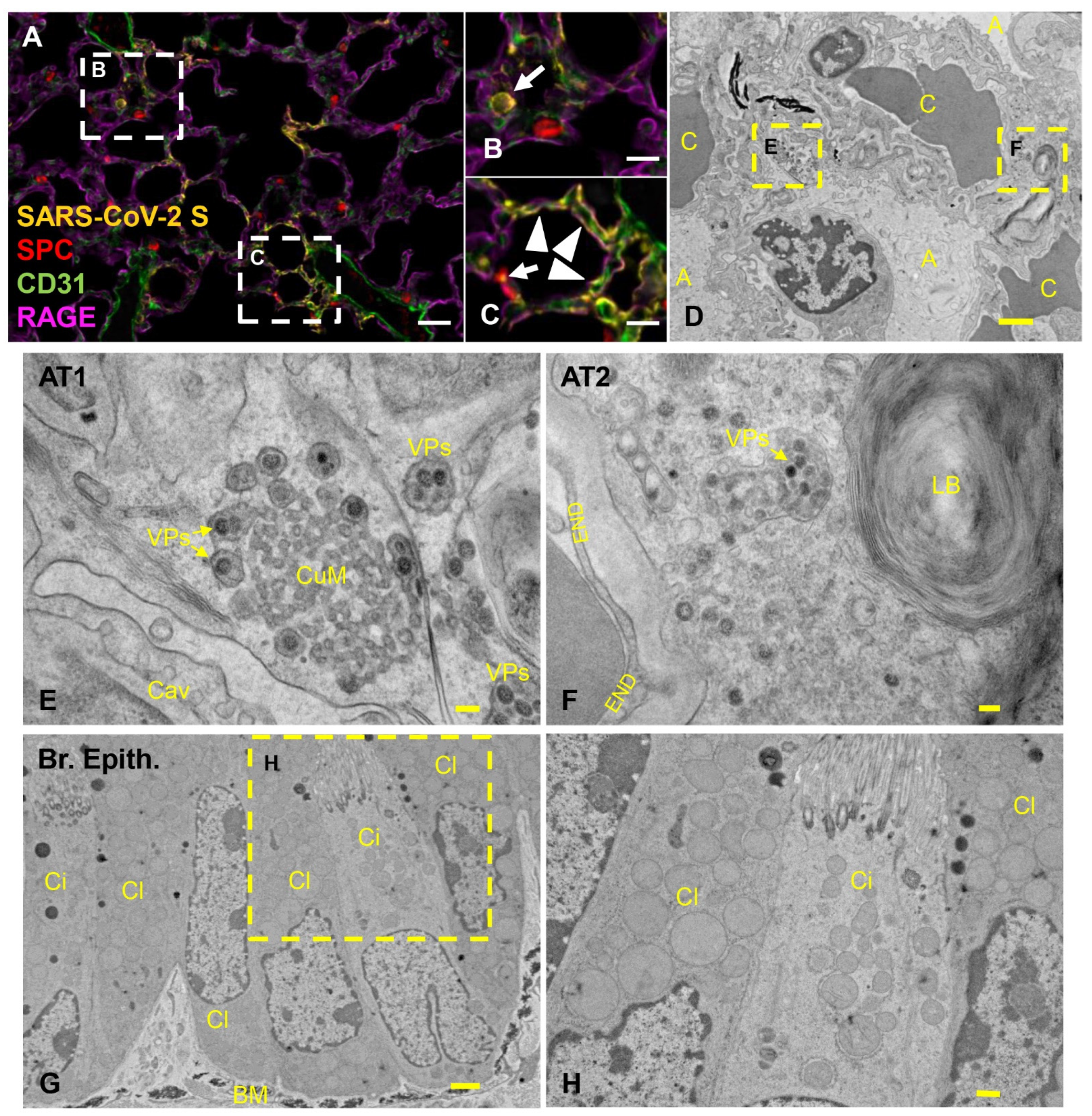
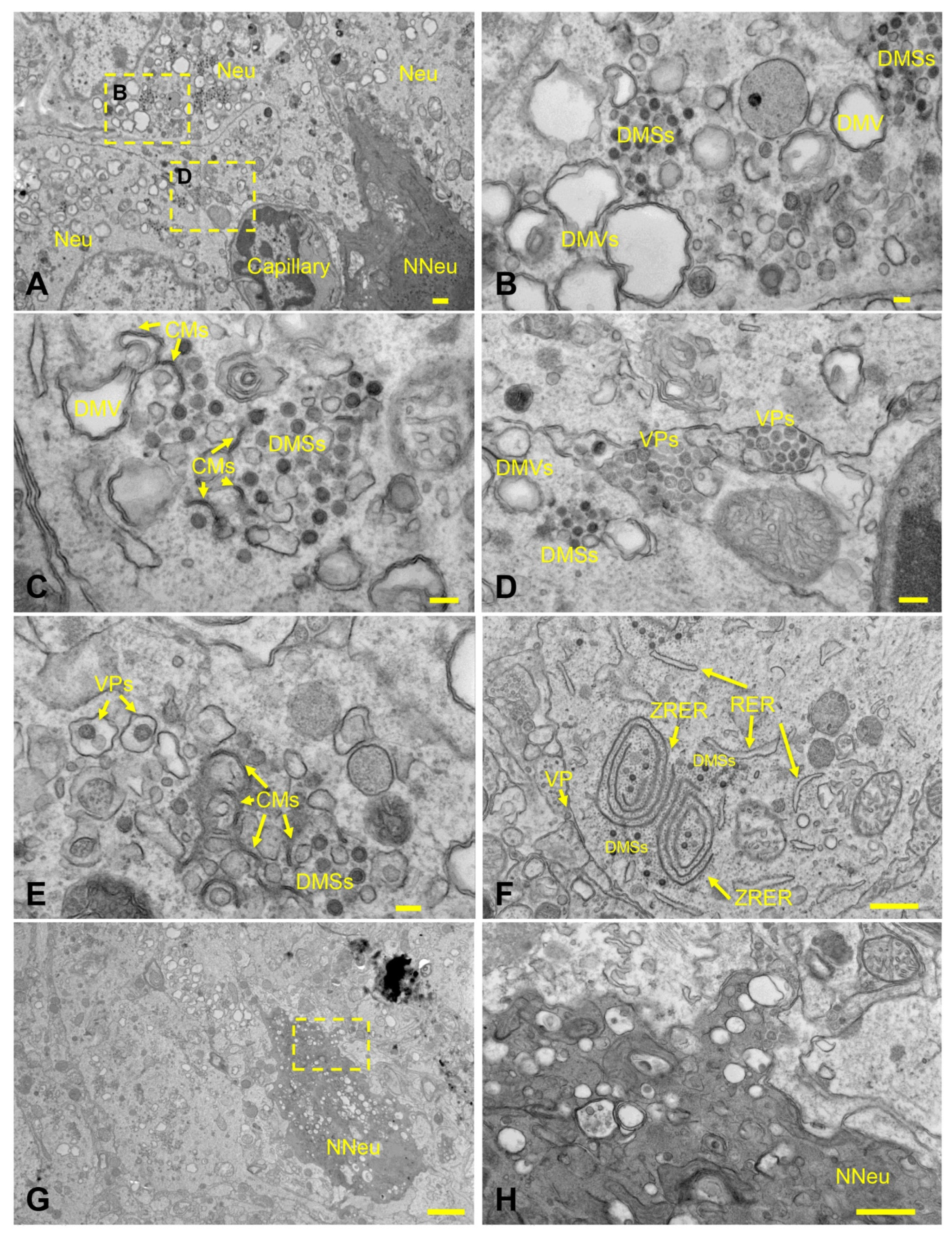
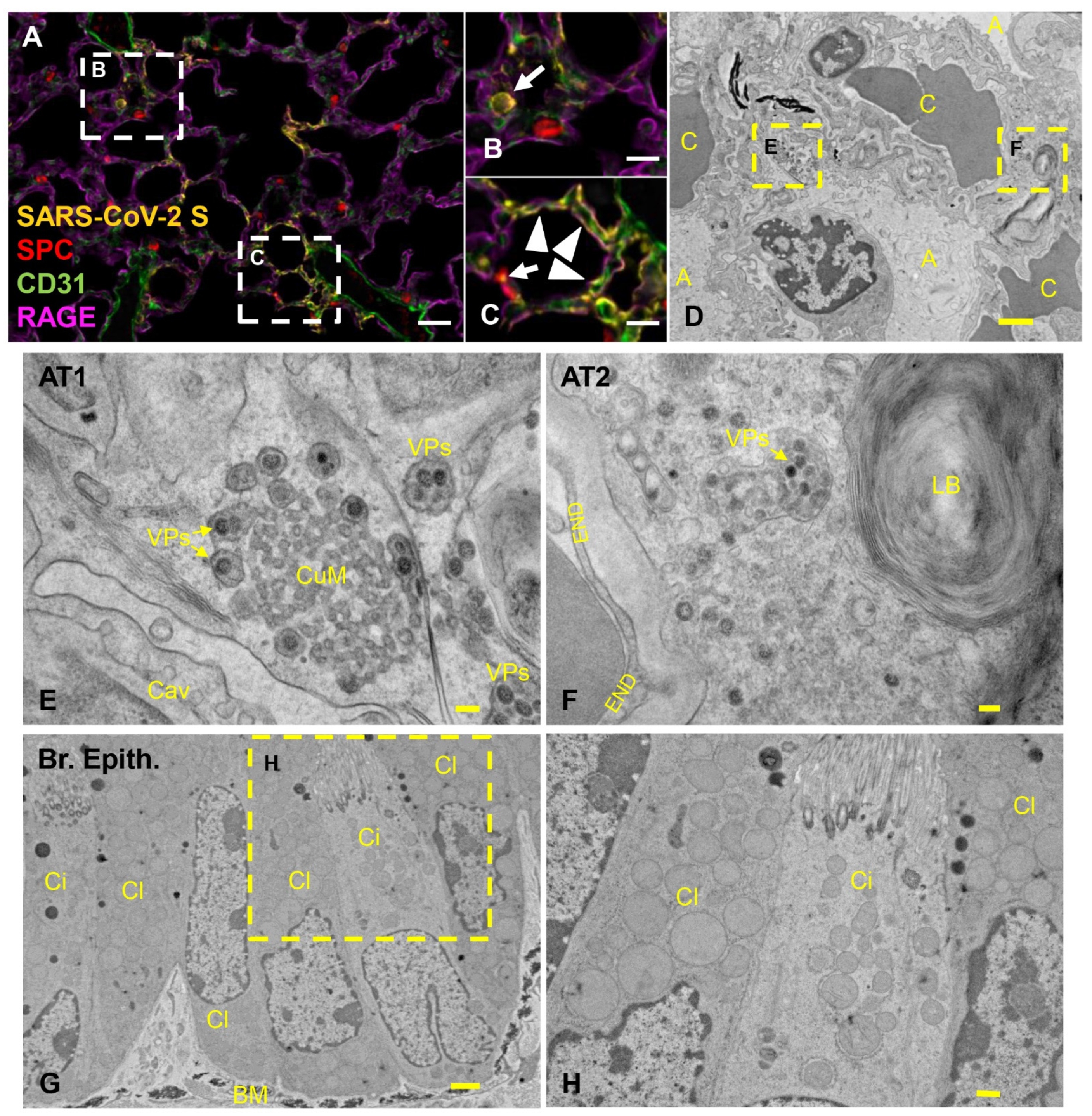
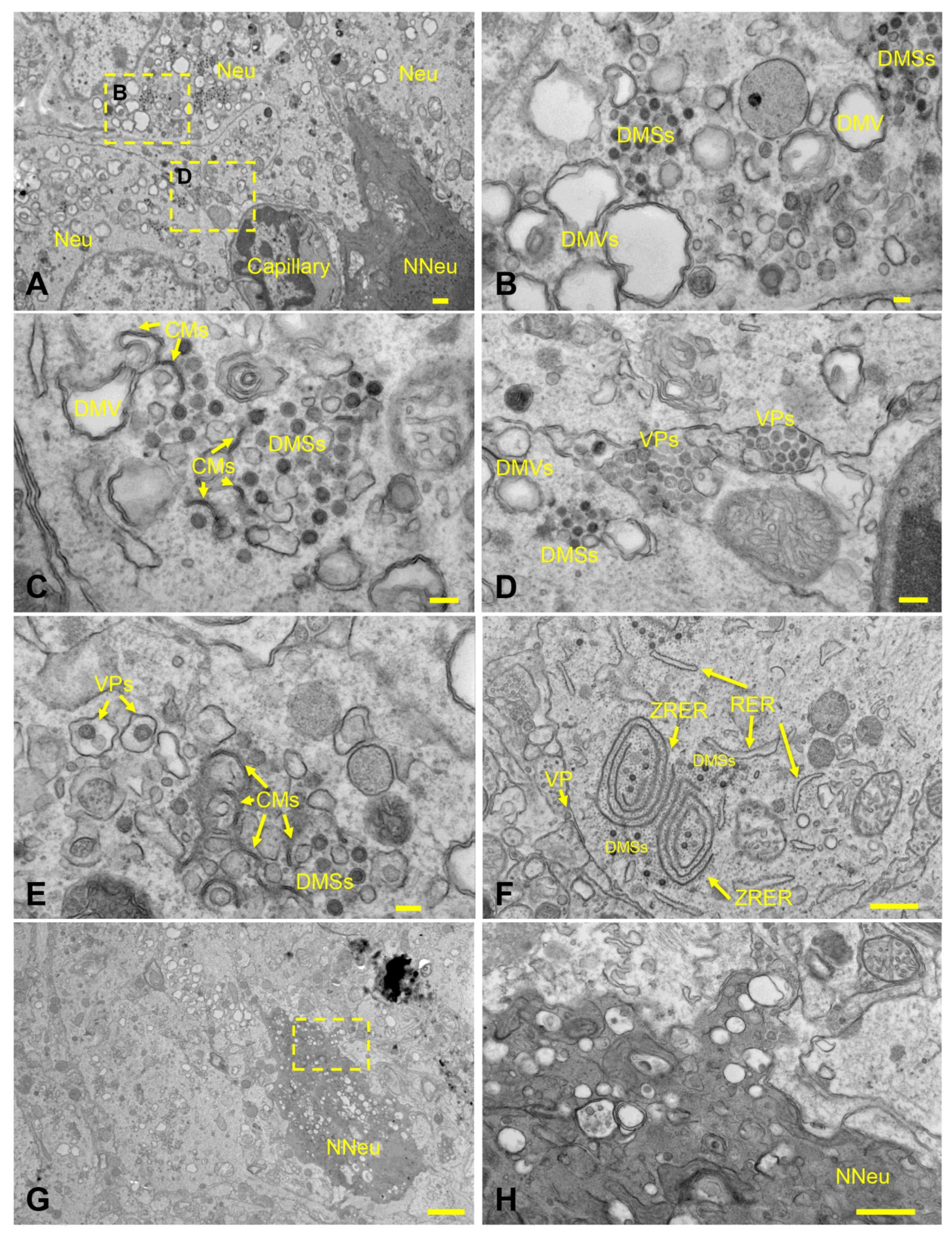
3.4. Pulmonary SARS-CoV-2 Replication and Assembly in K18-hACE2 Mice Occurs Exclusively in AT1 and AT2 Cells
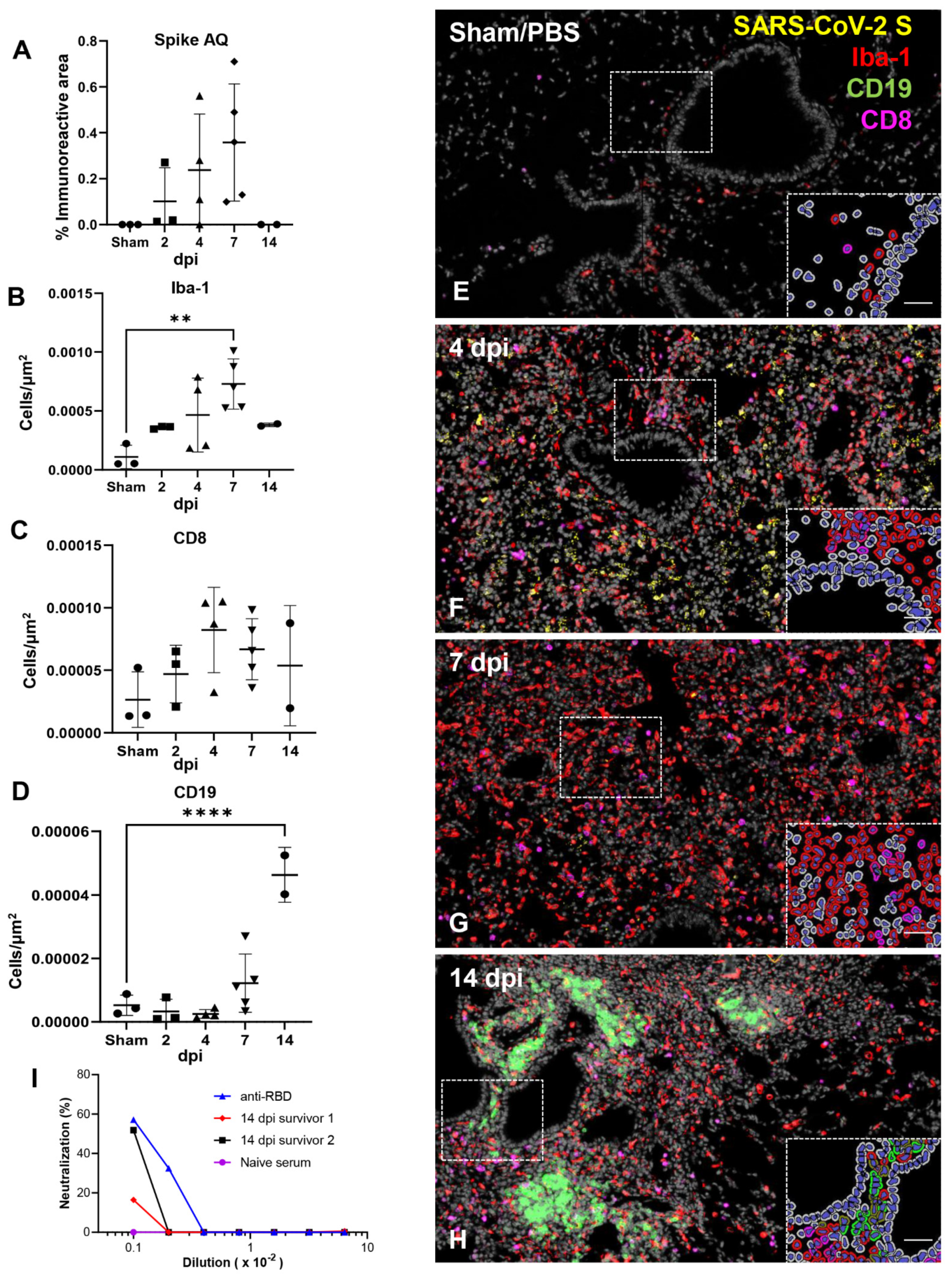
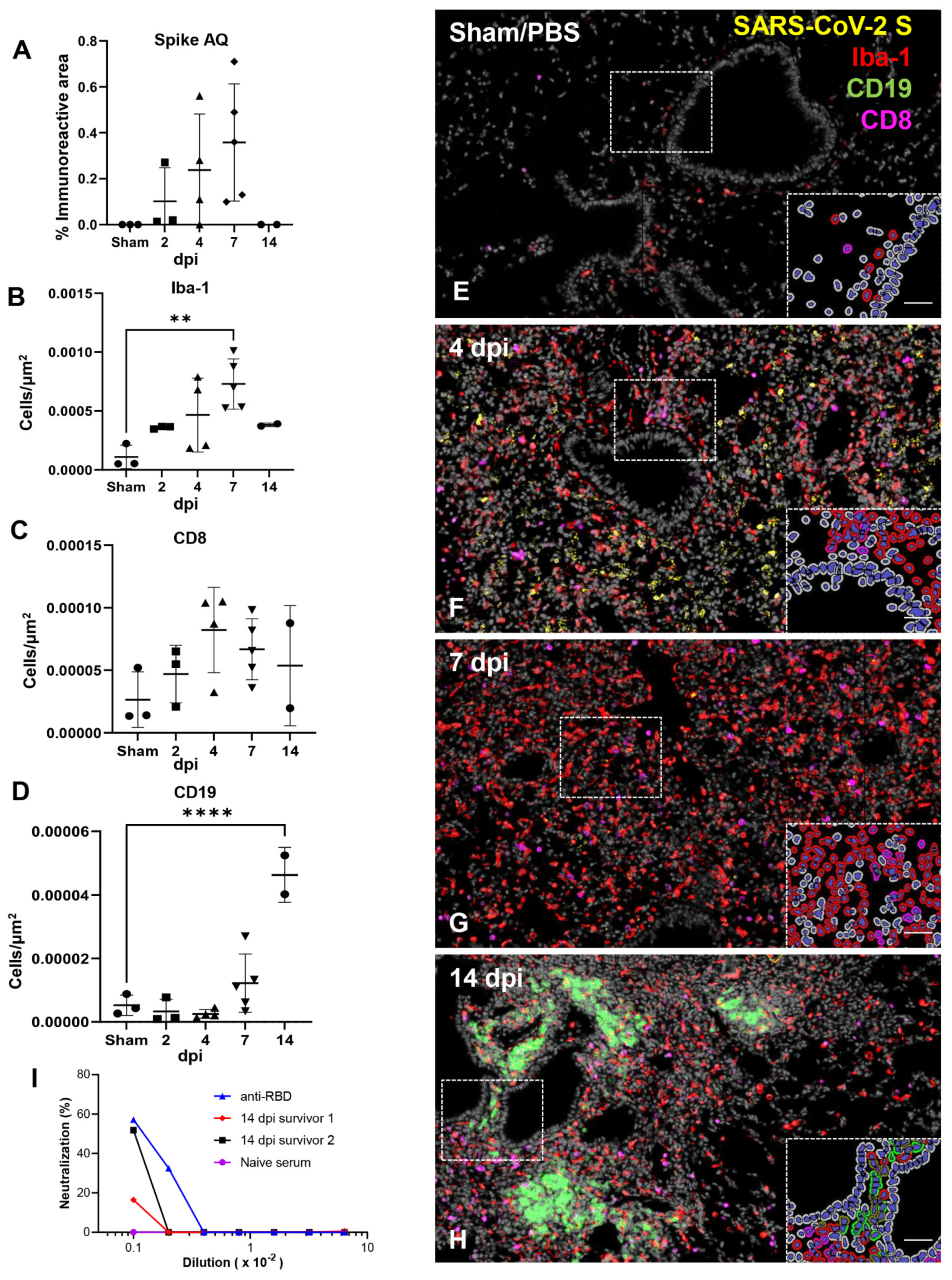
3.5. Effective Control of SARS-CoV-2 Infection in the Lower Respiratory Tract Is Associated with Recruitment of Macrophages and to a Lesser Degree Cytotoxic T Cells
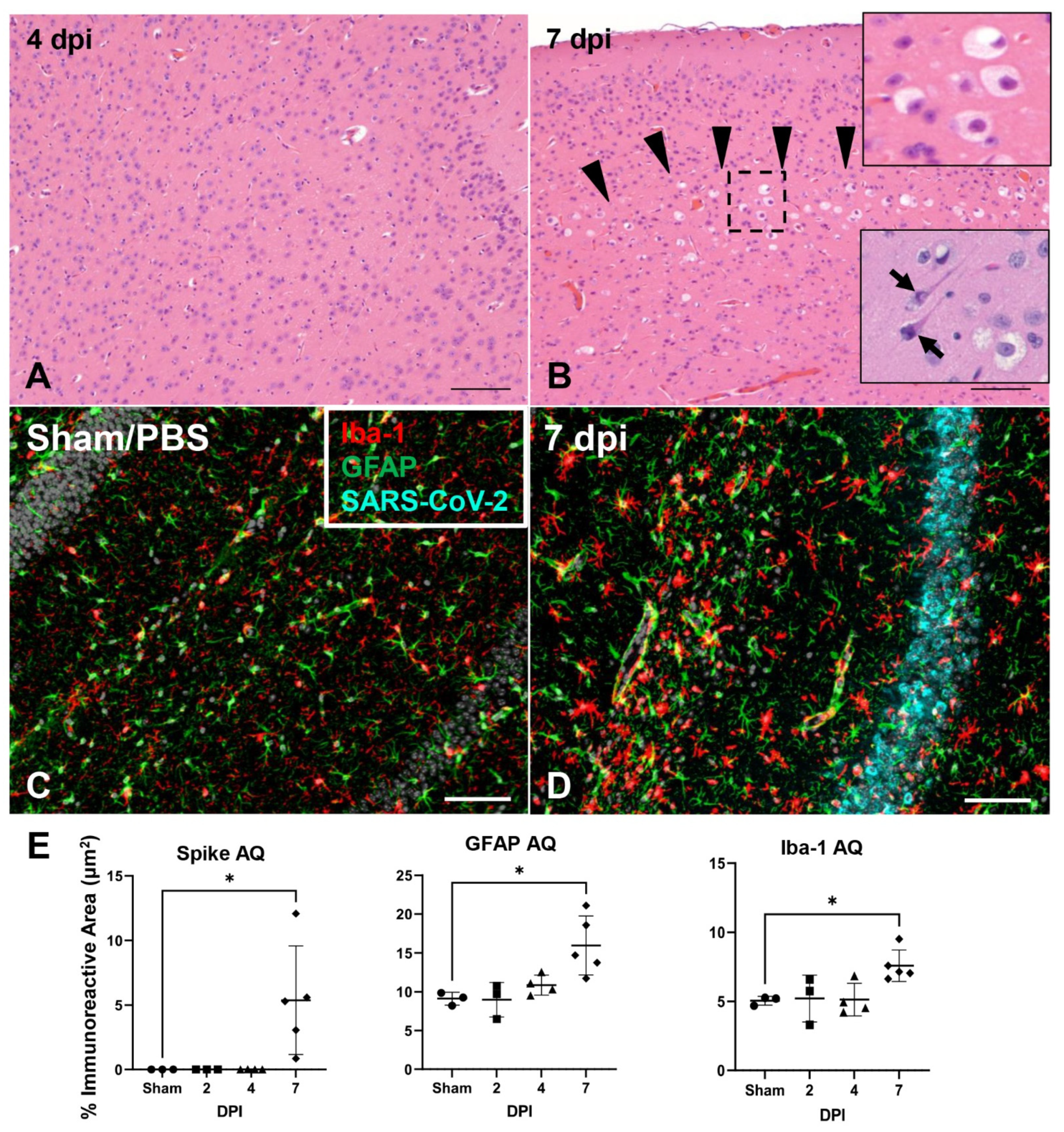
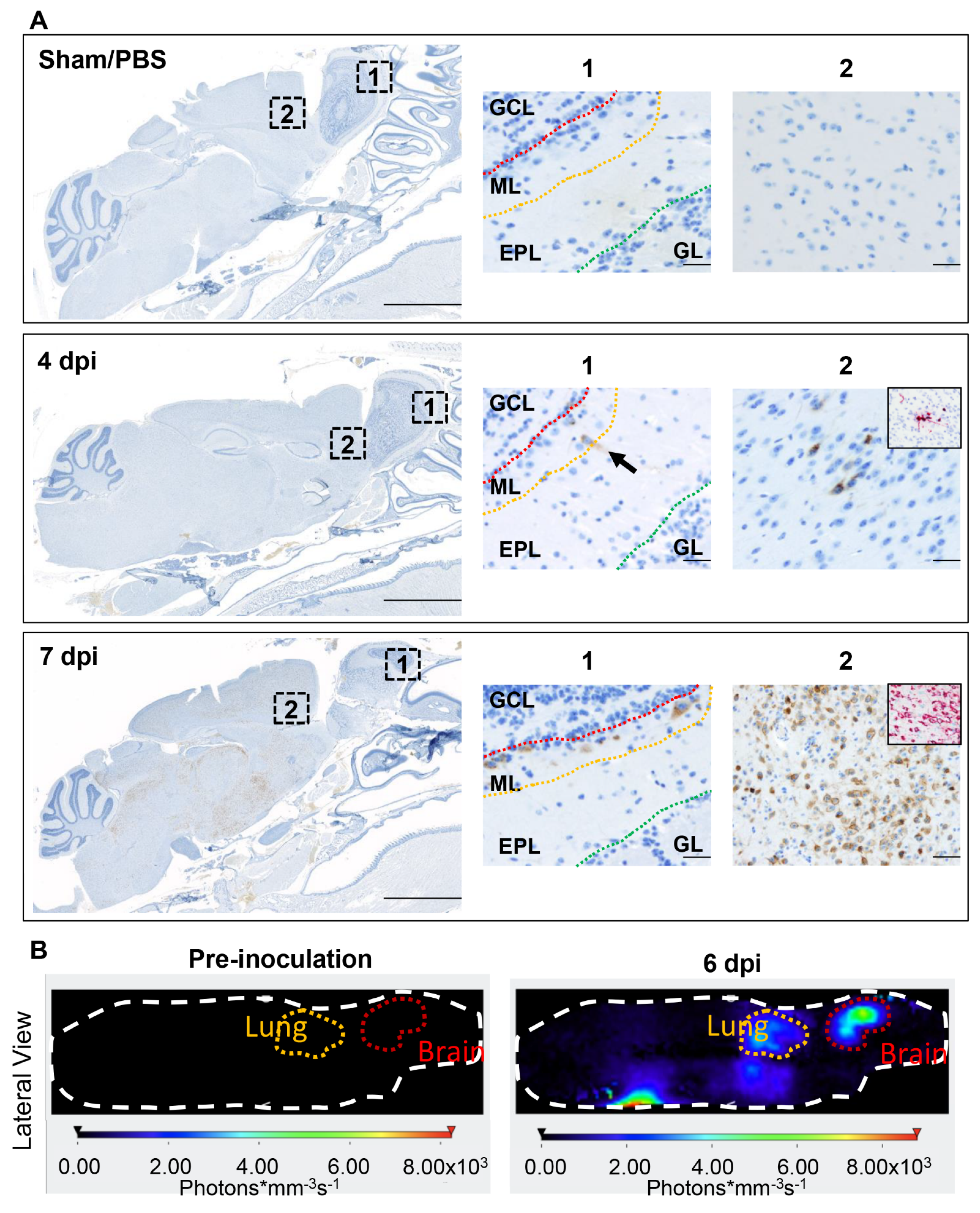
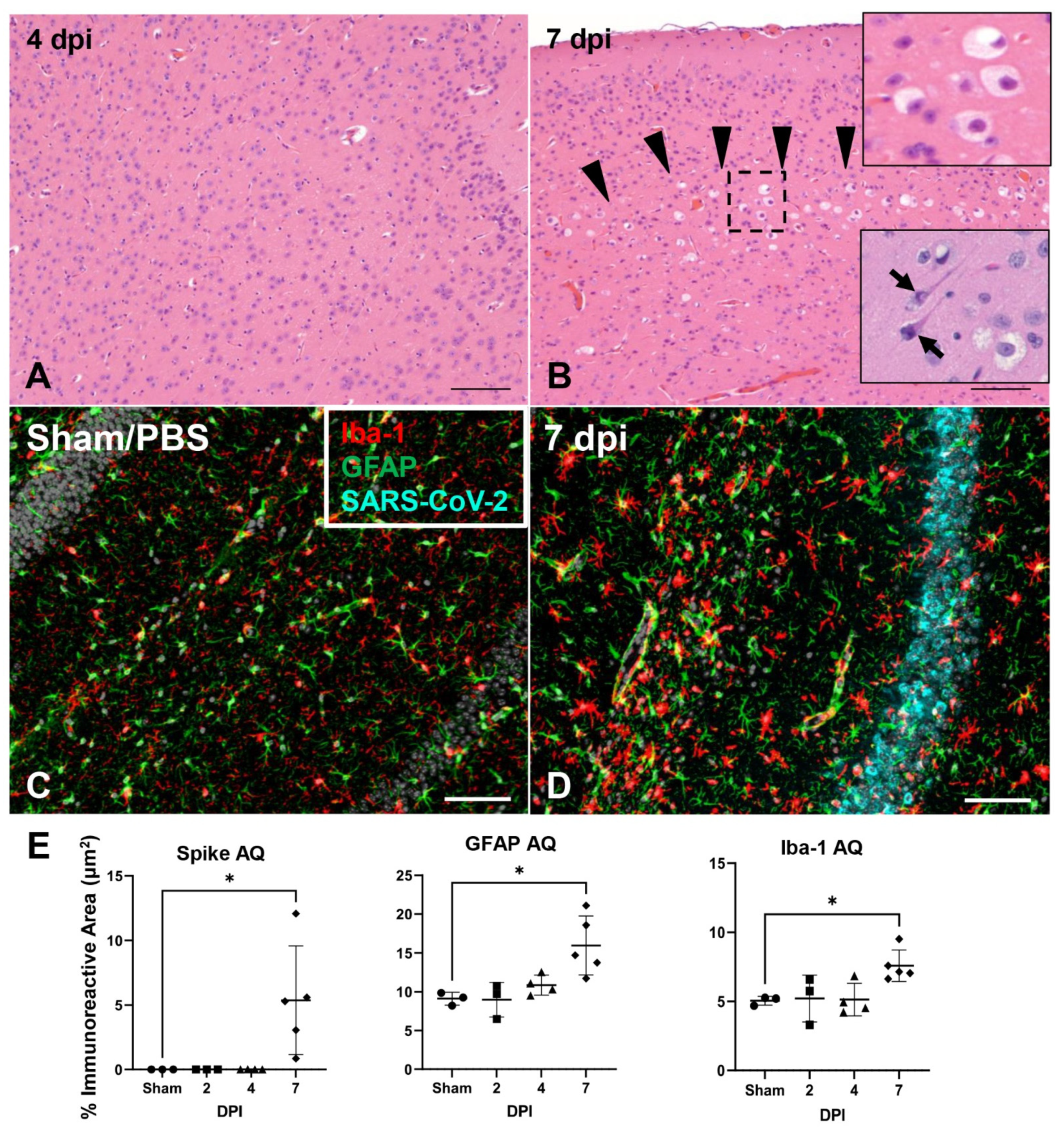
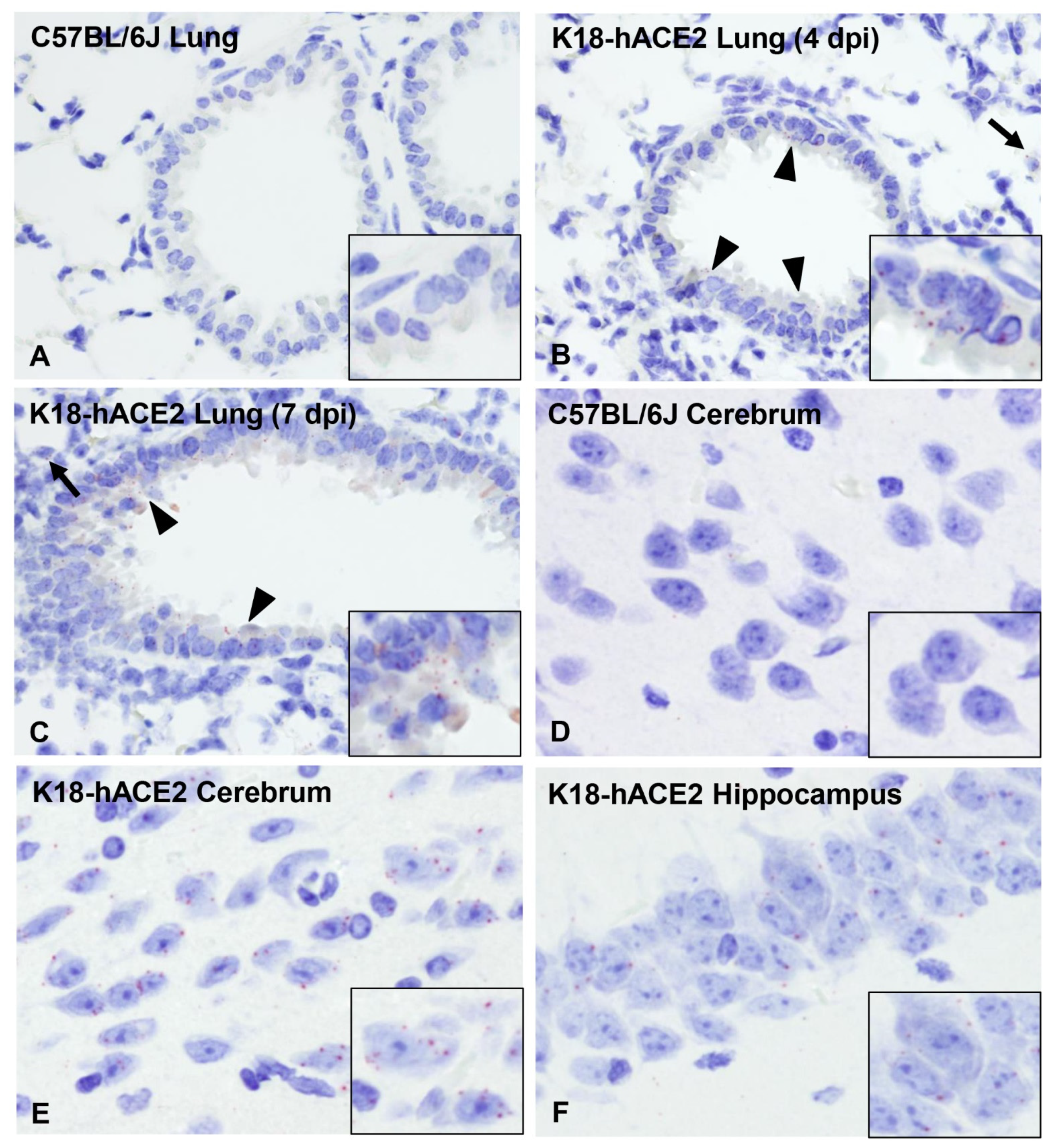
3.6. SARS-CoV-2 Exhibits Extensive Neuroinvasion with Resultant Neuronal Degeneration and Necrosis in K18-hACE2 Mice
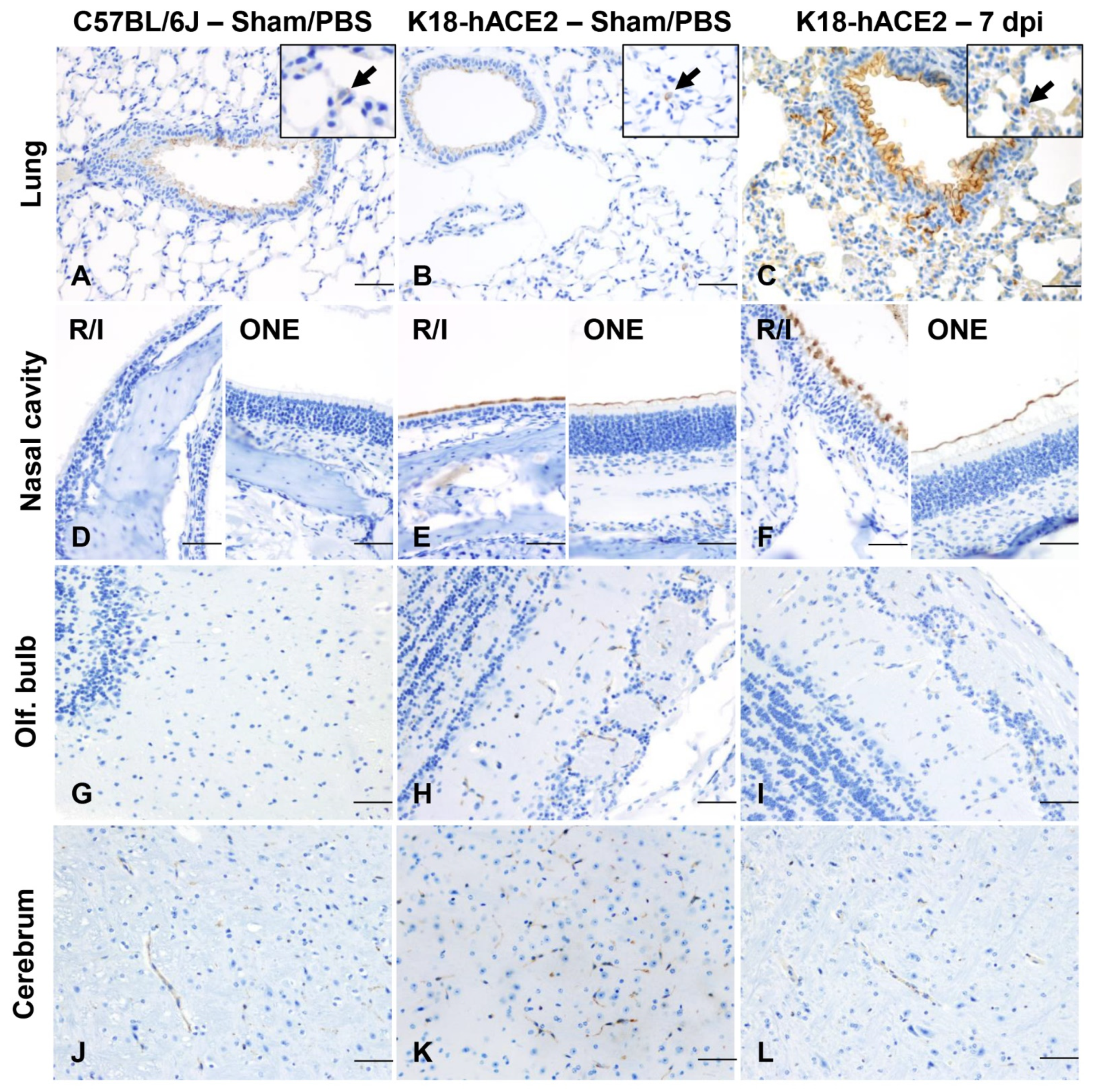
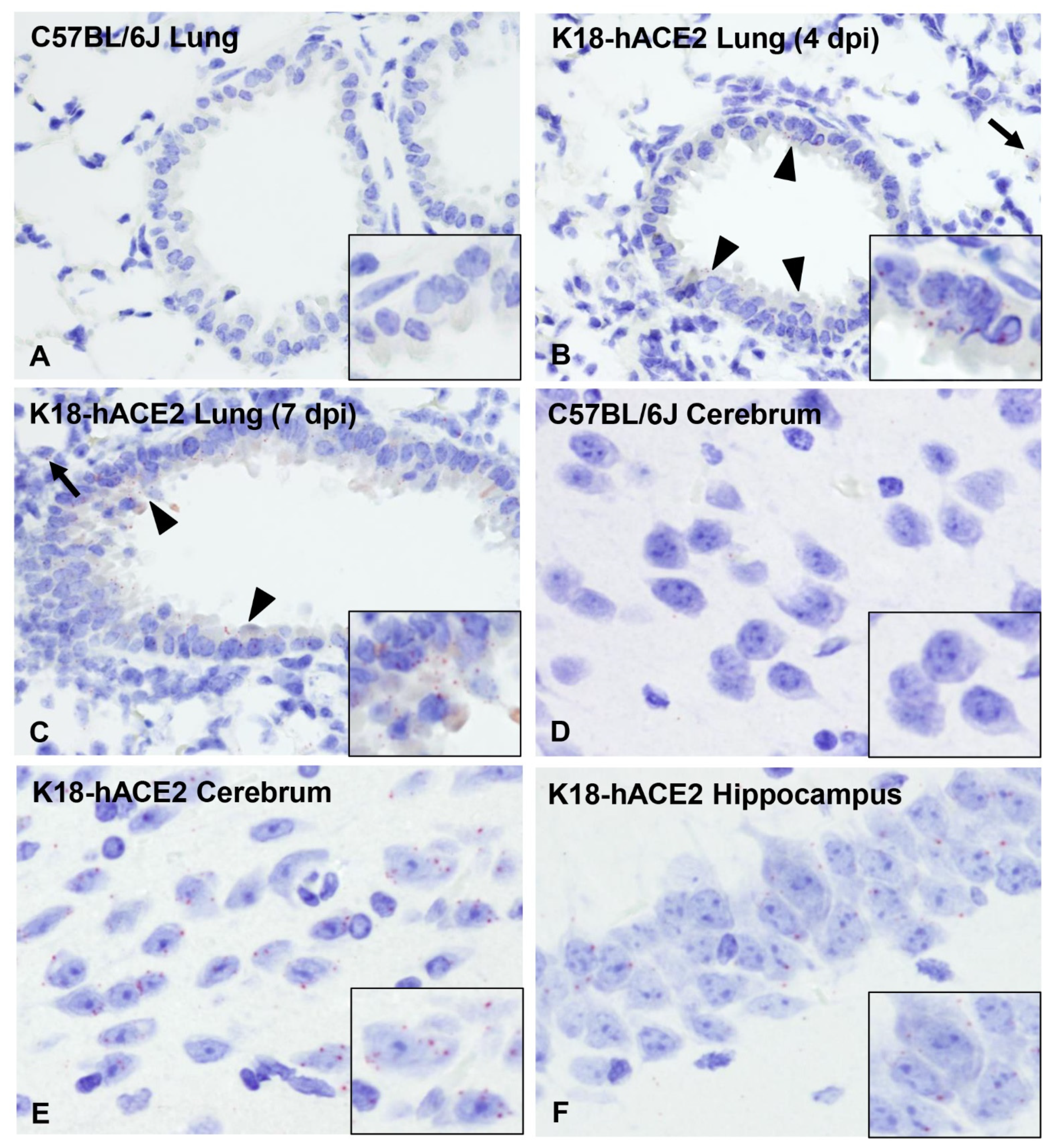
3.7. ACE2 Expression and Distribution Does Not Fully Reflect SARS-CoV-2 Cellular Tropism in K18-hACE2 Mice
3.8. Absence of Infection and Histologic Lesions in Extrapulmonary and Extraneural Tissues Despite ACE2 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Johns Hopkins University & Medicine: Coronavirus Resource Center. 2020. Available online: https://coronavirus.jhu.edu (accessed on 20 December 2021).
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R., Jr.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of COVID-19 in New York City. N. Engl. J. Med. 2020, 382, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Tenforde, M.W.; Kim, S.S.; Lindsell, C.J.; Billig Rose, E.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Erickson, H.L.; Steingrub, J.S.; Smithline, H.A.; et al. Symptom Duration and Risk Factors for Delayed Return to Usual Health among Outpatients with COVID-19 in a Multistate Health Care Systems Network—United States, March–June 2020. Morb. Mortal. Wkly. Rep. 2020, 69, 993–998. [Google Scholar] [CrossRef]
- Wang, F.; Kream, R.M.; Stefano, G.B. Long-Term Respiratory and Neurological Sequelae of COVID-19. Med. Sci. Monit. 2020, 26, e928996. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Tartof, S.Y.; Qian, L.; Hong, V.; Wei, R.; Nadjafi, R.F.; Fischer, H.; Li, Z.; Shaw, S.F.; Caparosa, S.L.; Nau, C.L.; et al. Obesity and Mortality among Patients Diagnosed with COVID-19: Results from an Integrated Health Care Organization. Ann. Intern. Med. 2020, 173, 773–781. [Google Scholar] [CrossRef]
- Team, C.C.-R. Preliminary Estimates of the Prevalence of Selected Underlying Health Conditions among Patients with Coronavirus Disease 2019—United States, 12 February–28 March 2020. Morb. Mortal. Wkly. Rep. 2020, 69, 382–386. [Google Scholar] [CrossRef]
- DosSantos, M.F.; Devalle, S.; Aran, V.; Capra, D.; Roque, N.R.; Coelho-Aguiar, J.D.M.; Spohr, T.; Subilhaga, J.G.; Pereira, C.M.; D’Andrea Meira, I.; et al. Neuromechanisms of SARS-CoV-2: A Review. Front. Neuroanat. 2020, 14, 37. [Google Scholar] [CrossRef]
- Eliezer, M.; Hamel, A.L.; Houdart, E.; Herman, P.; Housset, J.; Jourdaine, C.; Eloit, C.; Verillaud, B.; Hautefort, C. Loss of smell in patients with COVID-19: MRI data reveal a transient edema of the olfactory clefts. Neurology 2020, 95, e3145–e3152. [Google Scholar] [CrossRef] [PubMed]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with COVID-19. N. Engl. J. Med. 2020, 384, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Tan, B.H.; Wu, S.; Gui, Y.; Suo, J.L.; Li, Y.C. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J. Med. Virol. 2020, 93, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F.; Sabeti, P. Neuropathological Features of COVID-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Pottinger, G.; Scott, A.; Hopkins, C. Anosmia and loss of smell in the era of COVID-19. BMJ 2020, 370, m2808. [Google Scholar] [CrossRef]
- Johansen, M.D.; Irving, A.; Montagutelli, X.; Tate, M.D.; Rudloff, I.; Nold, M.F.; Hansbro, N.G.; Kim, R.Y.; Donovan, C.; Liu, G.; et al. Animal and translational models of SARS-CoV-2 infection and COVID-19. Mucosal. Immunol. 2020, 13, 877–891. [Google Scholar] [CrossRef]
- McNamara, T.; Richt, J.A.; Glickman, L. A Critical Needs Assessment for Research in Companion Animals and Livestock following the Pandemic of COVID-19 in Humans. Vector-Borne Zoonotic Dis. 2020, 20, 393–405. [Google Scholar] [CrossRef]
- Munoz-Fontela, C.; Dowling, W.E.; Funnell, S.G.P.; Gsell, P.S.; Riveros-Balta, A.X.; Albrecht, R.A.; Andersen, H.; Baric, R.S.; Carroll, M.W.; Cavaleri, M.; et al. Animal models for COVID-19. Nature 2020, 586, 509–515. [Google Scholar] [CrossRef]
- Gaudreault, N.; Trujillo, J.; Carossino, M.; Meekins, D.; Madden, D.; Balaraman, V.; Henningson, J.; Morozov, I.; Bold, D.; Kwong, T.; et al. SARS-CoV-2 infection, disease and transmission in domestic cats. Emerg. Microbes Inf. 2020, 9, 2322–2332. [Google Scholar] [CrossRef]
- Imai, M.; Iwatsuki-Horimoto, K.; Hatta, M.; Loeber, S.; Halfmann, P.J.; Nakajima, N.; Watanabe, T.; Ujie, M.; Takahashi, K.; Ito, M.; et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl. Acad. Sci. USA 2020, 117, 16587–16595. [Google Scholar] [CrossRef] [PubMed]
- Meekins, D.A.; Morozov, I.; Trujillo, J.D.; Gaudreault, N.N.; Bold, D.; Carossino, M.; Artiaga, B.L.; Indran, S.V.; Kwon, T.; Balaraman, V.; et al. Susceptibility of swine cells and domestic pigs to SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Oude Munnink, B.B.; de Meulder, D.; van Amerongen, G.; van den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, L.; Zhong, G.; Yuan, Q.; Wen, Z.; Wang, C.; He, X.; Liu, R.; Wang, J.; Zhao, Q.; Liu, Y.; et al. Replication, pathogenicity, and transmission of SARS-CoV-2 in minks. Natl. Sci. Rev. 2020, 8, nwaa291. [Google Scholar] [CrossRef] [PubMed]
- Sia, S.F.; Yan, L.M.; Chin, A.W.H.; Fung, K.; Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Conceicao, C.; Thakur, N.; Human, S.; Kelly, J.T.; Logan, L.; Bialy, D.; Bhat, S.; Stevenson-Leggett, P.; Zagrajek, A.K.; Hollinghurst, P.; et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020, 18, e3001016. [Google Scholar] [CrossRef]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.P.; Pfenning, A.R.; Zhao, H.; et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef]
- Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.D.; Liu, M.Q.; Chen, Y.; Shan, C.; Zhou, Y.W.; Shen, X.R.; Li, Q.; Zhang, L.; Zhu, Y.; Si, H.R.; et al. Pathogenesis of SARS-CoV-2 in Transgenic Mice Expressing Human Angiotensin-Converting Enzyme 2. Cell 2020, 182, 50–58.e8. [Google Scholar] [CrossRef] [PubMed]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathnasinghe, R.; Strohmeier, S.; Amanat, F.; Gillespie, V.L.; Krammer, F.; Garcia-Sastre, A.; Coughlan, L.; Schotsaert, M.; Uccellini, M.B. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. Emerg. Microbes Infect. 2020, 9, 2433–2445. [Google Scholar] [CrossRef]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2020, 589, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Chen, R.; He, X.; Yuan, Y.; Deng, X.; Li, R.; Yan, H.; Yan, S.; Liu, J.; Zhang, Y.; et al. Infection of wild-type mice by SARS-CoV-2 B.1.351 variant indicates a possible novel cross-species transmission route. Signal Transduct. Target. Ther. 2021, 6, 420. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.; Hu, B.; Chai, Y.; Yuen, T.T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022. [Google Scholar] [CrossRef]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5, e142032. [Google Scholar] [CrossRef]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.; Lopez-Figueroa, C.; Rodon, J.; Perez, M.; Brustolin, M.; Cantero, G.; Guallar, V.; Izquierdo-Useros, N.; Carrillo, J.; Blanco, J.; et al. Chronological brain lesions after SARS-CoV-2 infection in hACE2-transgenic mice. Vet. Pathol. 2021. [CrossRef]
- Fumagalli, V.; Rava, M.; Marotta, D.; Di Lucia, P.; Laura, C.; Sala, E.; Grillo, M.; Bono, E.; Giustini, L.; Perucchini, C.; et al. Administration of aerosolized SARS-CoV-2 to K18-hACE2 mice uncouples respiratory infection from fatal neuroinvasion. Sci. Immunol. 2021, 7, eabl9929. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.E.; Zhang, X.; Lokugamage, K.G.; Fontes-Garfias, C.R.; Zou, J.; Liu, J.; Ren, P.; Balakrishnan, M.; Cihlar, T.; et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 2020, 11, 5214. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martines, R.B.; Ritter, J.M.; Matkovic, E.; Gary, J.; Bollweg, B.C.; Bullock, H.; Goldsmith, C.S.; Silva-Flannery, L.; Seixas, J.N.; Reagan-Steiner, S.; et al. Pathology and Pathogenesis of SARS-CoV-2 Associated with Fatal Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 2005–2015. [Google Scholar] [CrossRef]
- Blair, R.V.; Vaccari, M.; Doyle-Meyers, L.A.; Roy, C.J.; Russell-Lodrigue, K.; Fahlberg, M.; Monjure, C.J.; Beddingfield, B.; Plante, K.S.; Plante, J.A.; et al. Acute Respiratory Distress in Aged, SARS-CoV-2-Infected African Green Monkeys but Not Rhesus Macaques. Am. J. Pathol. 2020, 191, 274–282. [Google Scholar] [CrossRef]
- Aid, M.; Busman-Sahay, K.; Vidal, S.J.; Maliga, Z.; Bondoc, S.; Starke, C.; Terry, M.; Jacobson, C.A.; Wrijil, L.; Ducat, S.; et al. Vascular Disease and Thrombosis in SARS-CoV-2-Infected Rhesus Macaques. Cell 2020, 183, 1354–1366.e13. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Barcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Moreau, G.B.; Burgess, S.L.; Sturek, J.M.; Donlan, A.N.; Petri, W.A.; Mann, B.J. Evaluation of K18-hACE2 Mice as a Model of SARS-CoV-2 Infection. Am. J. Trop. Med. Hyg. 2020, 103, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Oladunni, F.S.; Park, J.-G.; Tamayo, P.P.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin converting enzyme 2 transgenic mice. bioRxiv 2020. preprint. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Port, J.R.; Bushmaker, T.; Owusu, I.O.; Avanzato, V.A.; Fischer, R.J.; Schulz, J.E.; Holbrook, M.G.; Hebner, M.J.; Rosenke, R.; et al. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. bioRxiv 2020. preprint. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.H.; Chen, Q.; Gu, H.J.; Yang, G.; Wang, Y.X.; Huang, X.Y.; Liu, S.S.; Zhang, N.N.; Li, X.F.; Xiong, R.; et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe 2020, 28, 124–133.e4. [Google Scholar] [CrossRef]
- Leist, S.R.; Dinnon, K.H., 3rd; Schafer, A.; Tse, L.V.; Okuda, K.; Hou, Y.J.; West, A.; Edwards, C.E.; Sanders, W.; Fritch, E.J.; et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 2020, 183, 1070–1085.e12. [Google Scholar] [CrossRef]
- Maiese, A.; Manetti, A.C.; La Russa, R.; Di Paolo, M.; Turillazzi, E.; Frati, P.; Fineschi, V. Autopsy findings in COVID-19-related deaths: A literature review. Forensic Sci. Med. Pathol. 2020, 17, 279–296. [Google Scholar] [CrossRef]
- Bryche, B.; St Albin, A.; Murri, S.; Lacote, S.; Pulido, C.; Ar Gouilh, M.; Lesellier, S.; Servat, A.; Wasniewski, M.; Picard-Meyer, E.; et al. Massive transient damage of the olfactory epithelium associated with infection of sustentacular cells by SARS-CoV-2 in golden Syrian hamsters. Brain Behav. Immun. 2020, 89, 579–586. [Google Scholar] [CrossRef]
- Osterrieder, N.; Bertzbach, L.D.; Dietert, K.; Abdelgawad, A.; Vladimirova, D.; Kunec, D.; Hoffmann, D.; Beer, M.; Gruber, A.D.; Trimpert, J. Age-Dependent Progression of SARS-CoV-2 Infection in Syrian Hamsters. Viruses 2020, 12, 779. [Google Scholar] [CrossRef]
- Rosenke, K.; Meade-White, K.; Letko, M.; Clancy, C.; Hansen, F.; Liu, Y.; Okumura, A.; Tang-Huau, T.L.; Li, R.; Saturday, G.; et al. Defining the Syrian hamster as a highly susceptible preclinical model for SARS-CoV-2 infection. bioRxiv 2020, 9, 2673–2684. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Hysenaj, L.; Little, S.; Kulhanek, K.; Gbenedio, O.M.; Rodriguez, L.; Shen, A.; Lone, J.C.; Lupin-Jimenez, L.C.; Bonser, L.R.; Serwas, N.K.; et al. SARS-CoV-2 infection studies in lung organoids identify TSPAN8 as novel mediator. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, M.; Shen, W.; Rowan, N.R.; Kulaga, H.; Hillel, A.; Ramanathan, M., Jr.; Lane, A.P. Elevated ACE-2 expression in the olfactory neuroepithelium: Implications for anosmia and upper respiratory SARS-CoV-2 entry and replication. Eur. Respir. J. 2020, 56, 2001948. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2020, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Yoo, S.J.; Clijsters, M.; Backaert, W.; Vanstapel, A.; Speleman, K.; Lietaer, C.; Choi, S.; Hether, T.D.; Marcelis, L.; et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184, 5932–5949.e15. [Google Scholar] [CrossRef] [PubMed]
- Olivarria, G.M.; Cheng, Y.; Furman, S.; Pachow, C.; Hohsfield, L.A.; Smith-Geater, C.; Miramontes, R.; Wu, J.; Burns, M.S.; Tsourmas, K.I.; et al. Microglia do not restrict SARS-CoV-2 replication following infection of the central nervous system of K18-hACE2 transgenic mice. bioRxiv 2021, 96, jvi-01969. [Google Scholar] [CrossRef]
- Chen, R.E.; Winkler, E.S.; Case, J.B.; Aziati, I.D.; Bricker, T.L.; Joshi, A.; Darling, T.L.; Ying, B.; Errico, J.M.; Shrihari, S.; et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature 2021, 596, 103–108. [Google Scholar] [CrossRef]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Jager, M.; Desilets, A.; Olmstead, A.D.; Vandal, T.; Buchholz, D.W.; Imbiakha, B.; Gao, G.; et al. A novel highly potent inhibitor of TMPRSS2-like proteases blocks SARS-CoV-2 variants of concern and is broadly protective against infection and mortality in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Score = Criteria |
|---|---|
| Body weight | 1 = 10–19% loss |
| Respiration | 1 = rapid, shallow, increased effort |
| Appearance | 1 = ruffled fur, hunched posture |
| Responsiveness | 1 = low to moderate unresponsiveness |
| Neurologic signs | 1 = tremors |
| Sequence | Antigen Target | Species Origin | Clone | Manufacturer | Catalog | Primary Antibody Dilution | Antigen Retrieval (Ventana) | Chromogen or Fluorophore | |
|---|---|---|---|---|---|---|---|---|---|
| Assay 1 | NA | SARS-CoV-2 Spike (S) | Mouse | E7U6O | Cell Signaling Technology (Danvers, MA, USA) | Pre-commercialization | 1:1000 | CC1 (Tris) | DAB |
| Assay 2 | NA | Angiotensin converting enzyme 2 (ACE2) | Rabbit | EPR34435 | Abcam (Waltham, MA, USA) | ab108252 | 1:200 | CC1 (Tris) | Discovery Red and DAB |
| Assay 3 | NA | Neuropilin-1 | Rb | EPR3113 | Abcam | ab81321 | 1:50 | CC2 (Citrate) | DAB |
| Assay 4 | 1 | SARS-CoV-2 S | Mouse | E7U6O | Cell Signaling Technology | Pre-commercialization | 1:1000 | CC1 (Tris) | Opal 480 |
| 2 | Iba-1 | Rabbit | Polyclonal | WAKO (Richmond, VA, USA) | 019-19741 | 1:2000 | CC2 (Citrate) | Opal 570 | |
| 3 | GFAP | Rabbit | Polyclonal | DAKO (Glostrup, Denmark) | Z0334 | 1:500 | CC1 (Tris) | Opal 690 | |
| Assay 5 | 1 | CD8 | Rabbit | D4W2Z | Cell Signaling Technology | 98941 | 1:200 | CC1 (Tris) | Opal 620 |
| 2 | SARS-CoV-2 S | Mouse | E7U6O | Cell Signaling Technology | Pre-commercialization | 1:1000 | CC1 (Tris) | Opal 570 | |
| 3 | CD19 | Rabbit | D4V4B | Cell Signaling Technology | 90176 | 1:600 | CC2 (Citrate) | Opal 520 | |
| 4 | Iba-1 | Rabbit | Polyclonal | WAKO | 019-19741 | 1:2000 | CC2 (Citrate) | Opal 690 | |
| Assay 6 | 1 | RAGE | Rat | EPR21171 | R&D (Minneapolis, MN, USA) | MAB1179q-100 | 1:50 | CC1 (Tris) | Opal 480 |
| 2 | SARS-CoV N | Rabbit | Polyclonal | Novus biologicals (Littleton, CO, USA) | NB100-56576 | 1:200 | CC1 (Tris) | Opal 570 | |
| 3 | Prosurfactant C Protein | Rabbit | Polyclonal | Seven Hills Bioreagents (Cincinnati, Ohio, USA) | WRAB-9337 | 1:800 | CC2 (Citrate) | Opal 690 | |
| 4 | CD31 | Rabbit | D8V9E | Cell Signaling Technology | 77699S | 1:100 | CC2 (Citrate) | Opal 520 |
| DPI | AT1/AT2 | Bronchioles | Rostral Turbinates | Intermediate Turbinates | ONE | Olf. Bulb | Brain | Spinal Cord (CT) | Spinal Cord (LS) | GI * | Kidneys |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PBS/Sham (n = 3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 (n = 3) | 2 (1–2) | 0 | 1 (0–2) | 2 (1–2) | 1 (1–2) | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 (n = 5) | 2 (1–3) | 0 | 0 (0–1) | 0 (0–1) | 1 (0–1) | 0 (0–1) | 0 (0–1) | 0 | 0 | 0 | 0 |
| 6–8 (n = 13) | 2 (1–3) | 0 | 0 | 0 | 1 (0–1) | 1 (0–2) | 3 (0–3) | 1 (0–2) | 0 (0–1) | 0 | 0 |
| 14 (n = 2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carossino, M.; Kenney, D.; O’Connell, A.K.; Montanaro, P.; Tseng, A.E.; Gertje, H.P.; Grosz, K.A.; Ericsson, M.; Huber, B.R.; Kurnick, S.A.; et al. Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression. Viruses 2022, 14, 535. https://doi.org/10.3390/v14030535
Carossino M, Kenney D, O’Connell AK, Montanaro P, Tseng AE, Gertje HP, Grosz KA, Ericsson M, Huber BR, Kurnick SA, et al. Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression. Viruses. 2022; 14(3):535. https://doi.org/10.3390/v14030535
Chicago/Turabian StyleCarossino, Mariano, Devin Kenney, Aoife K. O’Connell, Paige Montanaro, Anna E. Tseng, Hans P. Gertje, Kyle A. Grosz, Maria Ericsson, Bertrand R. Huber, Susanna A. Kurnick, and et al. 2022. "Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression" Viruses 14, no. 3: 535. https://doi.org/10.3390/v14030535
APA StyleCarossino, M., Kenney, D., O’Connell, A. K., Montanaro, P., Tseng, A. E., Gertje, H. P., Grosz, K. A., Ericsson, M., Huber, B. R., Kurnick, S. A., Subramaniam, S., Kirkland, T. A., Walker, J. R., Francis, K. P., Klose, A. D., Paragas, N., Bosmann, M., Saeed, M., Balasuriya, U. B. R., ... Crossland, N. A. (2022). Fatal Neurodissemination and SARS-CoV-2 Tropism in K18-hACE2 Mice Is Only Partially Dependent on hACE2 Expression. Viruses, 14(3), 535. https://doi.org/10.3390/v14030535