
The Role of Nucleoprotein in Immunity to Human Negative-Stranded RNA Viruses—Not Just Another Brick in the Viral Nucleocapsid


Abstract
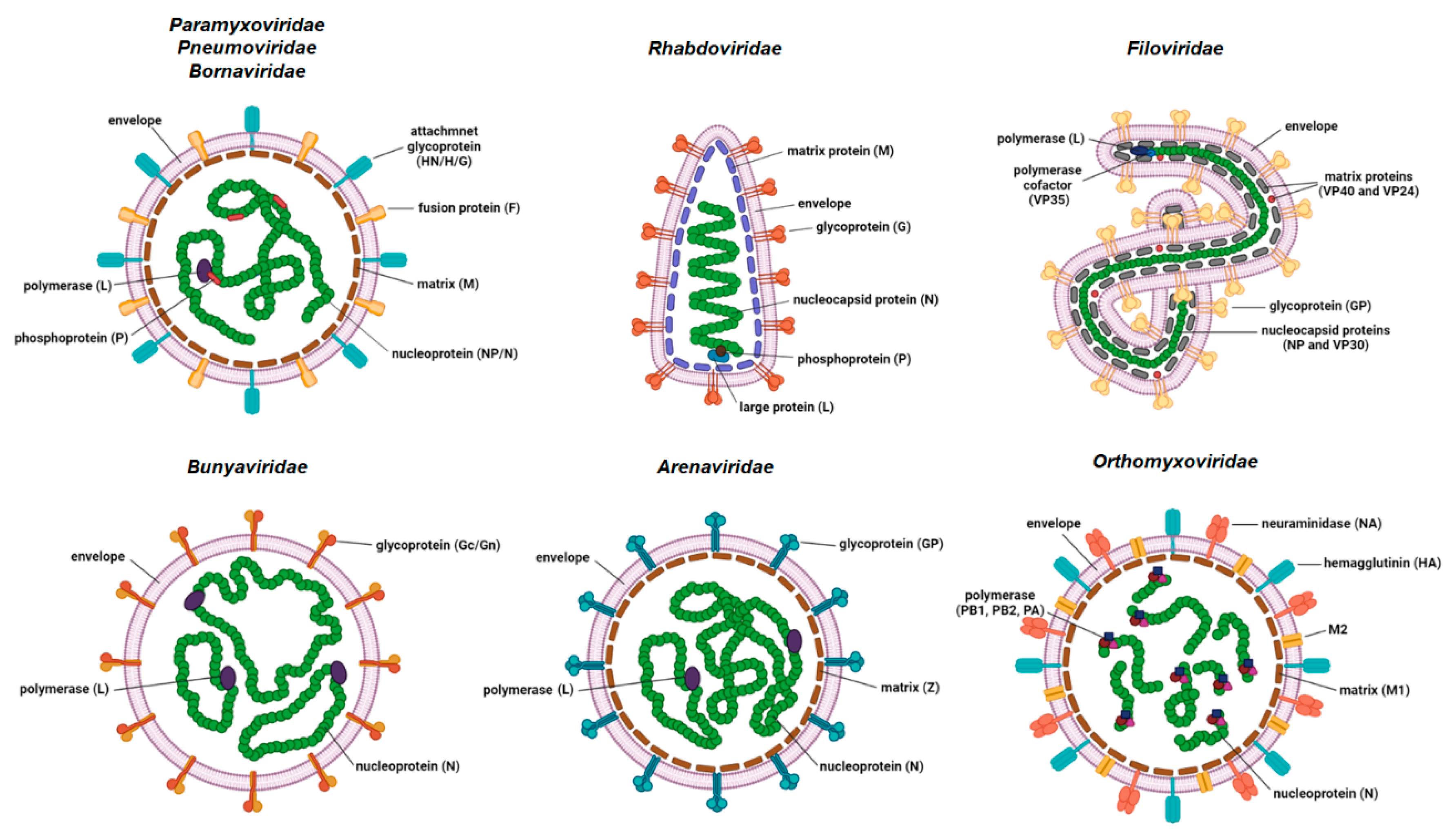
1. Introduction
2. Structure and Function of NP
2.1. Genetic Stability of NP
2.2. Architecture of NP and Structural Organization of NP–RNA Complex
2.3. Multiple Functions of NP
2.3.1. Protective Function
2.3.2. Role in Transcription and Replication
2.3.3. Immunosuppression
3. Immunity to NP
3.1. The Role of T-Cell Immune Response to NP
3.1.1. Paramyxoviruses: Measles and Mumps Viruses
3.1.2. Pneumoviruses: RSV and HMPV
3.1.3. Hantaviruses: Hantaan Virus
3.1.4. Filoviruses: Ebola Virus (EBOV)
3.1.5. Arenaviruses: Lassa Virus (LASV)
3.1.6. Orthomyxoviruses: Influenza Viruses

{kind=link}
{kind=link}
{kind=link}
| Location | Sequence | HLA Antigen 1 | Reference |
|---|---|---|---|
| 37–54 | GRFYIQMCTELKLSDYEG | A*01:01 A*02:01 B*08:01 | [175] |
| 39–47 | FYIQMCTEL | A*24:02 B*15:09 C*07:02 | [196] |
| 67–76 | RMVLSAFDER | A*03 | [174] |
| 91–99 | KTGGPIYRR | A*11:01 | [176] |
| 139–156 | WHSNLNDATYQRTRALVR | A*02:01 B*15:01 B*44:02 | [175] |
| 145–156 | DATYQRTRALVR | A*68:01 | [176] |
| 148–156 | TTYQRTRAL | A*02 | [174] |
| 158–166 | GMDPRMCSL | A*02 A*02:03 A*24:02 B*08:01 | [185,196] |
| 172–181 | LPRRSGAAGA | B*55:01 | [176] |
| 199–207 | RGINDRNFW | B*57:01 B*15:13 B*57:02 | [176,196] |
| 217–234 | IAYERMCNILKGKFQTAA | A*02:01 A*11:01 B*15:01 | [175] |
| 219–226 | YERMCNIL | B*:18:01 | [176] |
| 221–230 | RMCNILKGKF | B*44 | [174] |
| 225–233 | ILKGKFQTA | B*08:01 A*02:02 A*02:03 A*02:06 A*02:09 | [196] |
| 251–260 | AEIEDLIFLA | B*44 | [174] |
| 265–273 | ILRGSVAHK | A*03:01 A*02:03 A*11.01 A*33:01 A*68:01 | [176,182,185,196] |
| 328–336 | LVWMACHSA | A*02 | [185] |
| 329–339 | QLVWMACHSAA | A*02 | [174] |
| 331–348 | MACHSAAFELDRVLSFIK | A*02:01 A*24:02 B*12:02 B*35:03 | [175] |
| 335–349 | SAAFEDLRVLSFIKG | n.d. | [177] |
| 338–346 | FEDLRVLSF | B*37:01 | [174,176] |
| 379–395 | LELRSRYWAIRTRSGGN | A*01:01 A*02:01 B*08:01 B*07:02 | [175] |
| 380–388 | ELRSRYWAI | B*08:01 | [181] |
| 383–391 | SRYWAIRTR | B*27:05 | [176,182] |
| 397–414 | NQQRASAGQISIQPTFSV | A*02:01 A*11:01 B*15:01 B*44:02 | [175] |
| 418–426 | LPFEKSTVM | B*35:01 | [180] |
| 36–52 | IGRFYIQMCTELKLNDY | DR1 | [185] |
| 51–68 | DNEGRLIQNSLTIERMVL | DR1 | [185] |
| 75–89 | RNKYLEEHPSAGKDP | DR1 | [185] |
| 113–130 | KDEIRRIWRQANNGEDAT | DR1 | [185] |
| 147–154 | TYQRTRAL | DRB5*01:01 DRB1*07:01 DRB1*11:01 | [196] |
| 204–218 | RNFWRGENGRKTRSA | DR1 | [185] |
| 301–318 | IDPFRLLQNSQVYSLIRP | DR1 | [185] |
| 310–327 | SQVYSLIRPNENPAHKSQ | DR1 | [185] |
| 330–344 | LVWMACHSAAFEDLR | DR | [174] |
| 404–416 | GQISIQPTFSVQR | DRB1*04:04 | [184] |
| 409–425 | QPAFSVQRNLPFERVTI | DR1 | [185] |
| 463–475 | VFELSDEKAASPI | DRB1*09:01 | [184] |
3.2. B-Cell Response to NP
4. Vaccines Based on NP
4.1. Vaccine Candidates Based on the NP as Antigen in Clinical Trials
4.1.1. Influenza Vaccine Candidates Based on the NP
4.1.2. RSV Vaccine Candidates Based on the NP
4.2. Vaccines Based on NP as Antigen in Use
4.3. Nucleoprotein as a Scaffold for Foreign Antigen Delivery
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abraham, G.; Banerjee, A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1976, 73, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Wignall-Fleming, E.B.; Hughes, D.J.; Vattipally, S.; Modha, S.; Goodbourn, S.; Davison, A.J.; Randall, R.E. Analysis of Paramyxovirus Transcription and Replication by High-Throughput Sequencing. J. Virol. 2019, 93, e00571-19. [Google Scholar] [CrossRef] [PubMed]
- Emonet, S.F.; de la Torre, J.C.; Domingo, E.; Sevilla, N. Arenavirus genetic diversity and its biological implications. Infect. Genet Evolut. 2009, 9, 417–429. [Google Scholar] [CrossRef]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. After Marburg, Ebola. Lancet 1977, 1, 581–582. [Google Scholar]
- Hartman, A.L.; Towner, J.S.; Nichol, S.T. Ebola and marburg hemorrhagic fever. Clin. Lab. Med. 2010, 30, 161–177. [Google Scholar] [CrossRef]
- Laughlin, L.W.; Meegan, J.M.; Strausbaugh, L.J.; Morens, D.M.; Watten, R.H. Epidemic Rift Valley fever in Egypt: Observations of the spectrum of human illness. Trans. R. Soc. Trop. Med. Hyg. 1979, 73, 630–633. [Google Scholar] [CrossRef]
- Van Velden, D.J.J.; Meyer, J.D.; Oliver, J.; Gear, J.H.S.; McIntosh, B. Rift Valley fever affecting humans in South Africa. S. Afr. Med. J. 1977, 51, 867–871. [Google Scholar]
- Krüger, D.H.; Schönrich, G.; Klempa, B. Human pathogenic hantaviruses and prevention of infection. Hum. Vaccine 2011, 7, 685–693. [Google Scholar] [CrossRef]
- Monath, T.P. A short history of Lassa fever: The first 10–15 years after discovery. Curr. Opin. Virol. 2019, 37, 77–83. [Google Scholar] [CrossRef]
- Weiner, L.P.; Fleming, J.O. Viral infections of the nervous system. J. Neurosurg. 1984, 61, 207–224. [Google Scholar] [CrossRef]
- Tizard, I.; Ball, J.; Stoica, G.; Payne, S. The pathogenesis of bornaviral diseases in mammals. Anim. Health Res. Rev. 2016, 17, 92–109. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic Potential of Influenza A Viruses: A Comprehensive Overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, K.; Markowska-Daniel, I. In vivo reassortment of influenza viruses. Acta Biochim. Pol. 2014, 1, 427–431. [Google Scholar] [CrossRef]
- Beck, M.; Welsz-Malecek, R.; Mesko-Prejac, M.; Radman, V.; Juzbasic, M.; Rajninger-Miholic, M.; Prislin-Musklic, M.; Dobrovsak-Sourek, V.; Smerdel, S.; Stainer, D.W. Mumps vaccine L-Zagreb, prepared in chick fibroblasts. I. Production and field trials. J. Biol. Stand. 1989, 17, 85–90. [Google Scholar] [CrossRef]
- Buynak, E.B.; Hilleman, M.R. Live attenuated mumps virus vaccine. 1. Vaccine development. Proc. Soc. Exp. Biol. Med. 1966, 123, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Enders, J.F.; Katz, S.L.; Milovanovic, M.V.; Holloway, A. Studies of an attenuated measles virus vaccine: I. Development and preparation of the vaccine: Technics for assay of effects of vaccination. N. Engl. J. Med. 1960, 263, 153–159. [Google Scholar] [CrossRef]
- Ikić, D.; Juzbasić, M.; Beck, M.; Hrabar, A.; Cimbur-Schreiber, T. Attenuation and characterisation of Edmonston-Zagreb measles virus. Ann. Immunol. Hung. 1972, 16, 175–181. [Google Scholar]
- Popow-Kraupp, T.; Kunz, C.; Hofmann, H. Antibody response after application of a new live attenuated mumps vaccine (Pariorix) measured by enzyme-linked immunosorbent assay (ELISA). J. Med. Virol. 1982, 10, 119–129. [Google Scholar] [CrossRef]
- Šantak, M.; Kosutić-Gulija, T.; Tesović, G.; Ljubin-Sternak, S.; Gjenero-Margan, I.; Betica-Radić, L.; Forcić, D. Mumps virus strains isolated in Croatia in 1998 and 2005: Genotyping and putative antigenic relatedness to vaccine strains. J. Med. Virol. 2006, 78, 638–643. [Google Scholar] [CrossRef]
- Ivancic-Jelecki, J.; Santak, M.; Forcic, D. Variability of hemagglutinin-neuraminidase and nucleocapsid protein of vaccine and wild-type mumps virus strains. Infect. Genet. Evol. 2008, 8, 603–613. [Google Scholar] [CrossRef]
- Šantak, M.; Lang-Balija, M.; Ivancic-Jelecki, J.; Košutić-Gulija, T.; Ljubin-Sternak, S.; Forcic, D. Antigenic differences between vaccine and circulating wild-type mumps viruses decreases neutralization capacity of vaccine-induced antibodies. Epidemiol. Infect. 2013, 141, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Šantak, M.; Örvell, C.; Gulija, T.K. Identification of conformational neutralization sites on the fusion protein of mumps virus. J. Gen. Virol. 2015, 96, 982–990. [Google Scholar] [CrossRef] [PubMed]
- May, M.; Rieder, C.A.; Rowe, R.J. Emergent lineages of mumps virus suggest the need for a polyvalent vaccine. Int. J. Infect. Dis. 2018, 66, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Voigt, E.A.; Kennedy, R.B.; Poland, G.A. Knowledge gaps persist and hinder progress in eliminating mumps. Vaccine 2018, 36, 3721–3726. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, T.; Barbezange, C.; Francart, A.; Hamouda, A.; Litzroth, A.; Hutse, V.; Martens, L.; Vandermarliere, E.; Van Gucht, S. Sera from different age cohorts in Belgium show limited cross-neutralization between the mumps vaccine and outbreak strains. Clin. Microbiol. Infect. 2019, 25, 907. [Google Scholar] [CrossRef]
- Connell, A.R.; Connell, J.; Leahy, T.R.; Hassan, J. Mumps Outbreaks in Vaccinated Populations—Is It Time to Re-assess the Clinical Efficacy of Vaccines? Front. Immunol. 2020, 11, 2089. [Google Scholar] [CrossRef]
- Won, H.; Kim, A.R.; Yoo, J.S.; Chung, G.T.; Kang, H.J.; Kim, S.J.; Kim, S.S.; Lee, J.W. Cross-neutralization between vaccine and circulating wild-type mumps viruses in Korea. Vaccine 2021, 39, 1870–1876. [Google Scholar] [CrossRef]
- Šantak, M. Old and new ways to combat human influenza virus. Period. Biol. 2012, 114, 221–234. [Google Scholar]
- World Health Organization. Recommended Composition of Influenza Virus Vaccines for Use in the 2021–2022 Northern Hemisphere Influenza Season. Available online: https://www.who.int/publications/i/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2021-2022-northern-hemisphere-influenza-season (accessed on 13 January 2022).
- World Health Organization. Seasonal Influenza Vaccines: An Overview for Decision-Makers. Available online: https://apps.who.int/iris/handle/10665/336951 (accessed on 13 January 2022).
- Shu, L.L.; Bean, W.J.; Webster, R.G. Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J. Virol. 1993, 67, 2723–2729. [Google Scholar] [CrossRef]
- Šantak, M.; Mlinarić-Galinović, G.; Vilibić-Čavlek, T.; Tabain, I. Comparative genomics of human rubulavirus 2. Arch. Virol. 2018, 163, 3141–3148. [Google Scholar] [CrossRef]
- Örvell, C.; Rydbeck, R.; Löve, A. Immunological relationships between mumps virus and parainfluenza viruses studied with monoclonal antibodies. J. Gen. Virol. 1986, 67, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Tsurudome, M.; Nishio, M.; Komada, H.; Bando, H.; Ito, Y. Extensive antigenic diversity among human parainfluenza type 2 virus isolates and immunological relationships among paramyxoviruses revealed by monoclonal antibodies. Virology 1989, 171, 38–48. [Google Scholar] [CrossRef]
- Komada, H.; Klippmark, E.; Orvell, C.; Randall, R.E.; Ito, Y.; Norrby, E. Immunological relationships between parainfluenza virus type 4 and other paramyxoviruses studied by use of monoclonal antibodies. Arch. Virol. 1991, 116, 277–283. [Google Scholar] [CrossRef]
- Zhang, Y.; Pohl, J.; Brooks, W.A.; Erdman, D.D. Serologic cross-reactions between nucleocapsid proteins of human respiratory syncytial virus and human metapneumovirus. J. Clin. Microbiol. 2015, 53, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, Y.; Guo, Y.; Lou, Z. Structural perspective on the formation of ribonucleoprotein complex in negative-sense single-stranded RNA viruses. Trends Microbiol. 2013, 21, 475–484. [Google Scholar] [CrossRef]
- Luo, M.; Terrell, J.R.; McManus, S.A. Nucleocapsid Structure of Negative Strand RNA Virus. Viruses 2020, 12, 835. [Google Scholar] [CrossRef]
- Li, T.; Shen, Q.T. Insights into Paramyxovirus Nucleocapsids from Diverse Assemblies. Viruses 2021, 13, 2479. [Google Scholar] [CrossRef]
- Dong, S.; Yang, P.; Li, G.; Liu, B.; Wang, W.; Liu, X.; Xia, B.; Yang, C.; Lou, Z.; Guo, Y.; et al. Insight into the Ebola virus nucleocapsid assembly mechanism: Crystal structure of Ebola virus nucleoprotein core domain at 1.8 Å resolution. Protein Cell 2015, 6, 351–362. [Google Scholar] [CrossRef]
- Reguera, J.; Cusack, S.; Kolakofsky, D. Segmented negative strand RNA virus nucleoprotein structure. Curr. Opin. Virol. 2014, 5, 7–15. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Kraus, I.; Dickmanns, A.; Eickmann, M.; Garten, W.; Ficner, R. Crystal Structure of the Borna Disease Virus Nucleoprotein. Structure 2003, 11, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, Y.; Lou, Z. A versatile building block: The structures and functions of negative-sense single-stranded RNA virus nucleocapsid proteins. Protein Cell 2012, 3, 893–902. [Google Scholar] [CrossRef]
- Desfosses, A.; Goret, G.; Estrozi, L.F.; Ruigrok, R.W.H.; Gutsche, I. Nucleoprotein-RNA Orientation in the Measles Virus Nucleocapsid by Three-Dimensional Electron Microscopy. J. Virol. 2011, 85, 1391–1395. [Google Scholar] [CrossRef]
- Alayyoubi, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc. Natl. Acad. Sci. USA 2015, 112, E1792–E1799. [Google Scholar] [CrossRef]
- Murphy, S.K.; Parks, G.D. Genome nucleotide lengths that are divisible by six are not essential but enhance replication of defective interfering RNAs of the paramyxovirus simian virus 5. Virology 1997, 232, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Newcomb, W.W.; Brown, J.C.; Wall, J.S.; Hainfeld, J.F.; Trus, B.L.; Steven, A.C. Mass and molecular composition of vesicular stomatitis virus: A scanning transmission electron microscopy analysis. J. Virol. 1985, 54, 598–607. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Abelson, D.M.; Li, S.; Wood, M.R.; Saphire, E.O. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep. 2015, 12, 140–149. [Google Scholar] [CrossRef]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.; Jamin, M.; Ringkjøbing-Jensen, M.; et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef]
- Habchi, J.; Blangy, S.; Mamelli, L.; Ringkjobing, J.M.; Blackledge, M.; Darbon, H.; Oglesbee, M.; Shu, Y.; Longhi, S. Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipaviruses. J. Biol. Chem. 2011, 286, 13583–13602. [Google Scholar] [CrossRef]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Johansson, K.; Receveur-Bréchot, V.; Oldfield, C.J.; Dunker, A.K.; Canard, B.; Longhi, S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004, 99, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Habchi, J.; El Habre, Z.; Nesme, L.; Guigliarelli, B.; Belle, V.; Longhi, S. Assessing induced folding within the intrinsically disordered C-terminal domain of the Henipavirus nucleoproteins by site directed spin labeling EPR spectroscopy. J. Biomol. Struct. Dyn. 2013, 31, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Baronti, L.; Erales, E.; Habchi, J.; Felli, I.C.; Pierattelli, R.; Longhi, S. Dynamics of the intrinsically disordered C-terminal domain of the Nipah virus nucleoprotein and interaction with the X domain of the phosphoprotein as unveiled by NMR spectroscopy. ChemBioChem 2015, 16, 268–276. [Google Scholar] [CrossRef] [PubMed]
- D’Urzo, A.; Konijnenberg, A.; Rossetti, G.; Habchi, J.; Li, J.; Carloni, P.; Sobott, F.; Longhi, S.; Grandori, R. Molecular basis for structural heterogeneity of an intrinsically disordered protein bound to a partner by combined ESI-IM-MS and modeling. J. Am. Soc. Mass Spectrom. 2015, 26, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Su, X.; Li, T.; Qin, Y.; Zhang, N.; Yang, L.; Ma, L.; Bai, Y.; Qi, L.; Liu, Y.; et al. Structural plasticity of mumps virus nucleocapsids with cryo-EM structures. Commun. Biol. 2021, 4, 833. [Google Scholar] [CrossRef]
- Karlin, D.; Longhi, S.; Canard, B. Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology 2002, 302, 420–432. [Google Scholar] [CrossRef]
- Bhella, D.; Ralph, A.; Yeo, R.P. Conformational Flexibility in Recombinant Measles Virus Nucleocapsids Visualised by Cryo-negative Stain Electron Microscopy and Real-space Helical Reconstruction. J. Mol. Biol. 2004, 340, 319–331. [Google Scholar] [CrossRef]
- Schoehn, G.; Mavrakis, M.; Albertini, A.; Wade, R.; Hoenger, A.; Ruigrok, R.W.H. The 12 A° Structure of Trypsin-treated Measles Virus N–RNA. J. Mol. Biol. 2004, 339, 301–312. [Google Scholar] [CrossRef]
- Guryanov, S.G.; Liljeroos, L.; Kasaragod, P.; Kajander, T.; Butcher, S.J. Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J. Virol. 2016, 90, 2849–2857. [Google Scholar] [CrossRef]
- Albertini, A.A.V.; Wemimont, A.K.; Muziol, T.; Ravelli, R.B.G.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W.H. Crystal Structure of the Rabies Virus Nucleoprotein-RNA Complex. Science 2006, 313, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.H.; Crepin, T. Nucleoproteins of Negative Strand RNA Viruses; RNA Binding, Oligomerisation and Binding to Polymerase Co-Factor. Viruses 2010, 2, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagné, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Structural virology. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef]
- Cox, R.; Pickar, A.; Qiu, S.; Tsao, J.; Rodenburg, C.; Dokland, T.; Elson, A.; He, B.; Luo, M. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc. Natl. Acad. Sci. USA 2014, 111, 15208–15213. [Google Scholar] [CrossRef]
- Bharat, T.A.M.; Riches, J.D.; Kolesnikova, L.; Welsch, S.; Kraehling, V.; Davey, N.; Parsy, M.; Becker, S.; Briggs, J.A.G. Cryo-Electron Tomography of Marburg Virus Particles and Their Morphogenesis within Infected Cells. PLoS Biol. 2011, 9, e1001196. [Google Scholar] [CrossRef]
- Bharat, T.A.M.; Noda, T.; Riches, J.D.; Kraehling, V.; Kolesnikova, L.; Becker, S.; Kawaoka, Y.; Briggs, J.A.G. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4275–4280. [Google Scholar] [CrossRef]
- Ng, A.K.; Zhang, H.; Tan, K.; Li, Z.; Liu, J.; Chan, P.K.; Li, S.; Chan, W.; Au, S.W.; Joachimiak, A.; et al. Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008, 22, 3638–3647. [Google Scholar] [CrossRef]
- Raymond, D.D.; Piper, M.E.; Gerrard, S.R.; Smith, J.L. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. USA 2010, 107, 11769–11774. [Google Scholar] [CrossRef]
- Ferron, F.; Li, Z.; Danek, E.I.; Luo, D.; Wong, Y.; Coutard, B.; Lantez, V.; Charrel, R.; Canard, B.; Walz, T.; et al. The Hexamer Structure of the Rift Valley Fever Virus Nucleoprotein Suggests a Mechanism for its Assembly into Ribonucleoprotein Complexes. PLoS Pathog. 2011, 7, e1002030. [Google Scholar] [CrossRef]
- Olal, D.; Daumke, O. Structure of the Hantavirus Nucleoprotein Provides Insights into the Mechanism of RNA Encapsidation. Cell Rep. 2016, 14, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Houben, K.; Marion, D.; Tarbouriech, N.; Ruigrok, R.W.; Blanchard, L. Interaction of the C-terminal domains of sendai virus N and P proteins: Comparison of polymerase-nucleocapsid interactions within the paramyxovirus family. J. Virol. 2007, 81, 6807–6816. [Google Scholar] [CrossRef]
- Zheng, W.; Olson, J.; Vakharia, V.; Tao, Y.J. The crystal structure and RNA-binding of an orthomyxovirus nucleoprotein. PLoS Pathog. 2013, 9, e1003624. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.; Vasishtan, D.; Pražák, V.; Ghanem, A.; Conzelmann, K.K.; Rümenapf, T. Cryo EM structure of the rabies virus ribonucleoprotein complex. Sci. Rep. 2019, 9, 9639. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.S.; Xu, S.; Chen, Y.W.; Wang, J.H.; Shaw, P.C. Crystal structures of influenza nucleoprotein complexed with nucleic acid provide insights into the mechanism of RNA interaction. Nucleic Acids Res. 2021, 49, 4144–4154. [Google Scholar] [CrossRef] [PubMed]
- Chenavas, S.; Crépin, T.; Delmas, B.; Ruigrok, R.W.; Slama-Schwok, A. Influenza virus nucleoprotein: Structure, RNA binding, oligomerization and antiviral drug target. Future Microbiol. 2013, 8, 1537–1545. [Google Scholar] [CrossRef]
- Takaoka, A.; Yamada, T. Regulation of signaling mediated by nucleic acid sensors for innate interferon-mediated responses during viral infection. Int. Immunol. 2019, 31, 477–488. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Ortín, J.; Martín-Benito, J. The RNA synthesis machinery of negative-stranded RNA viruses. Virology 2015, 479–480, 532–544. [Google Scholar] [CrossRef]
- Qanungo, K.R.; Shaji, D.; Mathur, M.; Banerjee, A.K. Two RNA polymerase complexes from vesicular stomatitis virus-infected cells that carry out transcription and replication of genome RNA. Proc. Natl. Acad. Sci. USA 2004, 101, 5952–5957. [Google Scholar] [CrossRef]
- Blumberg, B.M.; Leppert, M.; Kolakofsky, D. Interaction of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell 1981, 23, 837–845. [Google Scholar] [CrossRef]
- Brunel, J.; Chopy, D.; Dosnon, M.; Bloyet, L.-M.; Devaux, P.; Urzua, E.; Cattaneo, R.; Longhi, S.; Gerlier, D. Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 2014, 88, 10851–10863. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Ueda, K.; Nagata, K.; Ishihama, A. RNA polymerase of influenza virus: Role of NP in RNA chain elongation. J. Biochem. 1988, 104, 1021–1026. [Google Scholar] [CrossRef]
- Turrell, L.; Lyall, J.W.; Tiley, L.S.; Fodor, E.; Vreede, F.T. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun. 2013, 4, 1591. [Google Scholar] [CrossRef]
- Hatakeyama, D.; Shoji, M.; Yamayoshi, S.; Yoh, R.; Ohmi, N.; Takenaka, S.; Saitoh, A.; Arakaki, Y.; Masuda, A.; Komatsu, T.; et al. Influenza A virus nucleoprotein is acetylated by histone acetyltransferases PCAF and GCN5. J. Biol. Chem. 2018, 293, 7126–7138. [Google Scholar] [CrossRef] [PubMed]
- Nilsson-Payant, B.E.; Blanco-Melo, D.; Uhl, S.; Escudero-Pérez, B.; Olschewski, S.; Thibault, P.; Panis, M.; Rosenthal, M.; Muñoz-Fontela, C.; Lee, B.; et al. Reduced Nucleoprotein Availability Impairs Negative-Sense RNA Virus Replication and Promotes Host Recognition. J. Virol. 2021, 95, e02274-20. [Google Scholar] [CrossRef]
- Yoshida, A.; Kawabata, R.; Honda, T.; Sakai, K.; Ami, Y.; Sakaguchi, T.; Irie, T. A Single Amino Acid Substitution within the Paramyxovirus Sendai Virus Nucleoprotein Is a Critical Determinant for Production of Interferon-Beta-Inducing Copyback-Type Defective Interfering Genomes. J. Virol. 2018, 92, e02094-17. [Google Scholar] [CrossRef]
- Tamashiro, V.G.; Perez, H.H.; Griffin, D.E. Prospective study of the magnitude and duration of changes in tuberculin reactivity during uncomplicated and complicated measles. Pediatr. Infect. Dis. J. 1987, 6, 451–454. [Google Scholar] [CrossRef]
- Hirsch, R.L.; Griffin, D.E.; Johnson, R.T.; Cooper, S.J.; de Soriano, I.L.; Roedenbeck, S.; Vaisberg, A. Cellular immune responses during complicated and uncomplicated measles virus infections of man. Clin. Immunol. Immunopathol. 1984, 31, 1–12. [Google Scholar] [CrossRef]
- Galama, J.M.D.; Ubels-Postma, J.; Vos, A.; Lucas, C.J. Measles virus inhibits acquisition of lymphocyte functions but not established effector functions. Cell. Immunol. 1980, 50, 401. [Google Scholar] [CrossRef]
- Griffin, D.E.; Ward, B.J. Differential CD4 T cell activation in measles. J. Infect. Dis. 1993, 168, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Esolen, L.M.; Ward, B.J.; Moench, T.R.; Griffin, D.E. Infection of monocytes during measles. J. Infect. Dis. 1993, 168, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ravanel, K.; Castelle, C.; Defrance, T.; Wild, T.F.; Charron, D.; Lotteau, V.; Rabourdin-Combe, C. Measles virus nucleocapsid protein binds to FcγRII and inhibits human B cell antibody production. J. Exp. Med. 1997, 186, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Kehren, J.; Trescol-Biemont, M.C.; Evlashev, A.; Valentin, H.; Walzer, T.; Tedone, R.; Loveland, B.; Nicolas, J.F.; Rabourdin-Combe, C.; et al. Mechanism of measles virus-induced suppression of inflammatory immune responses. Immunity 2001, 14, 69–79. [Google Scholar] [CrossRef]
- Marie, J.C.; Saltel, F.; Escola, J.M.; Jurdic, P.; Wild, T.F.; Horvat, B. Cell surface delivery of the measles virus nucleoprotein: A viral strategy to induce immunosuppression. J. Virol. 2004, 78, 11952–11961. [Google Scholar] [CrossRef]
- Laine, D.; Trescol-Biémont, M.C.; Longhi, S.; Libeau, G.; Marie, J.C.; Vidalain, P.O.; Azocar, O.; Diallo, A.; Canard, B.; Rabourdin-Combe, C.; et al. Measles virus (MV) nucleoprotein binds to a novel cell surface receptor distinct from FcgammaRII via its C-terminal domain: Role in MV-induced immunosuppression. J. Virol. 2003, 77, 11332–11346. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNAspecific 30 to 50 exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef]
- Russier, M.; Reynard, S.; Carnec, X.; Baize, S. The exonuclease domain of Lassa virus nucleoprotein is involved in antigen-presenting-cell-mediated NK cell responses. J. Virol. 2014, 88, 13811–13820. [Google Scholar] [CrossRef]
- Wolff, S.; Becker, S.; Groseth, A. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J. Virol. 2013, 87, 224–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, K.; Tripathi, S.; Ranjan, P.; Kumar, P.; Garten, R.; Deyde, V.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; et al. Influenza A virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS ONE 2011, 6, e20215. [Google Scholar] [CrossRef] [PubMed]
- Masatani, T.; Ito, N.; Shimizu, K.; Ito, Y.; Nakagawa, K.; Sawaki, Y.; Koyama, H.; Sugiyama, M. Rabies virus nucleoprotein functions to evade activation of the RIG-I-mediated antiviral response. J. Virol. 2010, 84, 4002–4012. [Google Scholar] [CrossRef] [PubMed]
- Masatani, T.; Ito, N.; Shimizu, K.; Ito, Y.; Nakagawa, K.; Abe, M.; Yamaoka, S.; Sugiyama, M. Amino acids at positions 273 and 394 in rabies virus nucleoprotein are important for both evasion of host RIG-I-mediated antiviral response and pathogenicity. Virus Res. 2011, 155, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Masatani, T.; Ito, N.; Ito, Y.; Nakagawa, K.; Abe, M.; Yamaoka, S.; Okadera, K.; Sugiyama, M. Importance of rabies virus nucleoprotein in viral evasion of interferon response in the brain. Microbiol. Immunol. 2013, 57, 511–517. [Google Scholar] [CrossRef]
- Song, W.; Kao, W.; Zhai, A.; Qian, J.; Li, Y.; Zhang, Q.; Zhao, H.; Hu, Y.; Li, H.; Zhang, F. Borna disease virus nucleoprotein inhibits type I interferon induction through the interferon regulatory factor 7 pathway. Biochem. Biophys. Res. Commun. 2013, 438, 619–623. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Soghoian, D.Z.; Streeck, H. Cytolytic CD4(+) T cells in viral immunity. Expert Rev. Vaccines 2010, 9, 1453–1463. [Google Scholar] [CrossRef]
- Lamb, J.R.; Eckels, D.D.; Lake, P.; Johnson, A.H.; Hartzman, R.J.; Woody, J.N. Antigen-specific human T lymphocyte clones: Induction, antigen specificity, and MHC restriction of influenza virus-immune clones. J. Immunol. 1982, 128, 233–238. [Google Scholar]
- Beňová, K.; Hancková, M.; Koči, K.; Kúdelová, M.; Betáková, T. T cells and their function in the immune response to viruses. Acta Virol. 2020, 64, 131–143. [Google Scholar] [CrossRef]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Jameson, J.; Cruz, J.; Ennis, F.A. Human cytotoxic T-lymphocyte repertoire to influenza A viruses. J. Virol. 1998, 72, 8682–8689. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.; Bellini, W. Measles virus. In Fields Virology, 3rd ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott–Raven: Philadelphia, PA, USA, 1996; pp. 1267–1298. [Google Scholar]
- Nanan, R.; Carstens, C.; Kreth, H.W. Demonstration of virus-specific CD8+ memory T cells in measles-seropositive individuals by in vitro peptide stimulation. Clin. Exp. Immunol. 1995, 102, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Kreth, H.W.; ter Meulen, V.; Eckert, G. Demonstration of HLA restricted cells in patients with acute measles. Med. Microbiol. Immunol. 1979, 165, 203–214. [Google Scholar] [CrossRef]
- Lucas, C.J.; Biddison, W.E.; Nelson, D.L.; Shaw, S. Killing of measles virus–infected cells by human cytotoxic T cells. Infect. Immun. 1982, 38, 226–232. [Google Scholar] [CrossRef]
- Sethi, K.K.; Stroehmann, I.; Brandis, H. Generation of cytolytic T-cell cultures displaying measles virus specificity and human histocompatibility leucyte antigen restriction. Infect. Immun. 1982, 36, 657–661. [Google Scholar] [CrossRef]
- Jacobson, S.; Richert, J.R.; Biddison, W.E.; Satinsky, A.; Hartzman, R.J.; McFarland, H.F. Measles virus–specific T4+ human cytotoxic T cell clones are restricted by class II HLA antigens. J. Immunol. 1984, 133, 754–757. [Google Scholar]
- Richert, J.R.; McFarland, H.F.; McFarlin, D.E.; Johnson, A.H.; Woody, J.H.; Hartzman, R.J. Measles-specific T cell clones derived from a twin with multiple sclerosis: Genetic restriction studies. J. Immunol. 1985, 34, 1561–1566. [Google Scholar]
- Jacobson, S.; Sekaly, R.P.; Jacobson, C.L.; McFarland, H.F.; Long, E.O. HLA class II–restricted presentation of cytoplasmic measles virus antigens to cytotoxic T cells. J. Virol. 1989, 63, 1756–1762. [Google Scholar] [CrossRef]
- van Binnendijk, R.S.; Poelen, M.C.; Kuijpers, K.C.; Osterhaus, A.D.; Uytdehaag, F.G. The predominance of CD8+ T cells after infection with measles virus suggests a role for CD8+ class I MHC-restricted cytotoxic T lymphocytes (CTL) in recovery from measles. Clonal analyses of human CD8+ class I MHC-restricted CTL. J. Immunol. 1990, 144, 2394–2399. [Google Scholar] [PubMed]
- Jaye, A.; Magnusen, A.F.; Whittle, H.C. Human leukocyte antigen class I- and class II-restricted cytotoxic T lymphocyte responses to measles antigens in immune adults. J. Infect. Dis. 1998, 177, 1282–1289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ilonen, J.; Mäkelä, M.J.; Ziola, B.; Salmi, A.A. Cloning of human T cell specific for measles virus haemagglutinin and nucleocapsid. Clin. Exp. Immunol. 1990, 81, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.; Rose, J.W.; Flerlage, M.L.; McFarlin, D.E.; McFarland, H.F. Induction of measles virus-specific human cytotoxic T cells by purified measles virus nucleocapsid and hemagglutinin polypeptides. Viral Immunol. 1987, 1, 153–162. [Google Scholar] [CrossRef]
- Hickman, C.J.; Khan, A.S.; Rota, P.A.; Bellini, W.J. Use of synthetic peptides to identify measles nucleoprotein T-cell epitopes in vaccinated and naturally infected humans. Virology 1997, 235, 386–397. [Google Scholar] [CrossRef]
- Marttila, J.; Ilonen, J.; Norrby, E.; Salmi, A. Characterization of T cell epitopes in measles virus nucleoprotein. J. Gen. Virol. 1999, 80, 1609–1615. [Google Scholar] [CrossRef][Green Version]
- Demotz, S.; Ammerlaan, W.; Fournier, P.; Muller, C.P.; Barbey, C. Processing of the DRB1*1103-restricted measles virus nucleoprotein determinant 185-199 in the endosomal compartment. Clin. Exp. Immunol. 1998, 114, 228–235. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Kenneth, L.; Johnson, K.L.; Muddiman, D.C.; Vierkant, R.A.; Poland, G.A. Identification and Characterization of Novel, Naturally Processed Measles Virus Class II HLA-DRB1 Peptides. J. Virol. 2004, 78, 42–51. [Google Scholar] [CrossRef][Green Version]
- Marshall, H.S.; Plotkin, S. The changing epidemiology of mumps in a high vaccination era. Lancet Infect. Dis. 2019, 19, 118–119. [Google Scholar] [CrossRef]
- de Wit, J.; Emmelot, M.E.; Meiring, H.; van Gaans-van den Brink, J.A.M.; van Els, C.A.C.M.; Kaaijk, P. Identification of Naturally Processed Mumps Virus Epitopes by Mass Spectrometry: Confirmation of Multiple CD8+ T-Cell Responses in Mumps Patients. J. Infect. Dis. 2020, 221, 474–482. [Google Scholar] [CrossRef]
- de Wit, J.; Emmelot, M.E.; Poelen, M.C.M.; Lanfermeijer, J.; Han, W.G.H.; van Els, C.A.C.M.; Kaaijk, P. The Human CD4(+) T Cell Response against Mumps Virus Targets a Broadly Recognized Nucleoprotein Epitope. J. Virol. 2019, 93, e01883-18. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, A.H.; Neijens, H.J.; Osterhaus, A.D. Pathogenesis of RSV lower respiratory tract infection: Implications for vaccine development. Vaccine 2001, 19, 2769–2782. [Google Scholar] [CrossRef]
- Johnson, K.M.; Bloom, H.H.; Mufson, M.A.; Chanock, R.M. Natural reinfection of adults by respiratory syncytial virus. Possible relation to mild upper respiratory disease. N. Engl. J. Med. 1962, 267, 68–72. [Google Scholar] [CrossRef]
- Hall, C.B.; Walsh, E.E.; Long, C.E.; Schnabel, K.C. Immunity to and frequency of reinfection with respiratory syncytial virus. J. Infect. Dis. 1991, 163, 693–698. [Google Scholar] [CrossRef]
- Ohuma, E.O.; Okiro, E.A.; Ochola, R.; Sande, C.J.; Cane, P.A.; Medley, G.F.; Bottomley, C.; Nokes, D.J. The natural history of respiratory syncytial virus in a birth cohort: The influence of age and previous infection on reinfection and disease. Am. J. Epidemiol. 2012, 176, 794–802. [Google Scholar] [CrossRef]
- Fishaut, M.; Tubergen, D.; McIntosh, K. Cellular response to respiratory viruses with particular reference to children with disorders of cell-mediated immunity. J. Pediatr. 1980, 96, 179–186. [Google Scholar] [CrossRef]
- Bangham, C.R.; McMichael, A.J. Specific human cytotoxic T cells recognize B-cell lines persistently infected with respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 1986, 83, 9183–9187. [Google Scholar] [CrossRef]
- Openshaw, P.J.; Chiu, C. Protective and dysregulated T cell immunity in RSV infection. Curr. Opin. Virol. 2013, 3, 468–474. [Google Scholar] [CrossRef]
- Lampert, P.W.; Joseph, B.S.; Oldstone, M.B. Antibody-induced capping of measles virus antigens on plasma membrane studied by electron microscopy. J. Virol. 1975, 15, 1248–1255. [Google Scholar] [CrossRef]
- González, A.E.; Lay, M.K.; Jara, E.L.; Espinoza, J.A.; Gómez, R.S.; Soto, J.; Rivera, C.A.; Abarca, K.; Bueno, S.M.; Riedel, C.A.; et al. Aberrant T cell immunity triggered by human Respiratory Syncytial Virus and human Metapneumovirus infection. Virulence 2017, 8, 685–704. [Google Scholar] [CrossRef]
- Bangham, C.R.; Openshaw, P.J.; Ball, L.A.; King, A.M.; Wertz, G.W.; Askonas, B.A. Human and murine cytotoxic T cells specific to respiratory syncytial virus recognize the viral nucleoprotein (N), but not the major glycoprotein (G), expressed by vaccinia virus recombinants. J. Immunol. 1986, 137, 3973–3977. [Google Scholar] [PubMed]
- Goulder, P.J.; Lechner, F.; Klenerman, P.; McIntosh, K.; Walker, B.D. Characterization of a novel respiratory syncytial virus-specific human cytotoxic T-lymphocyte epitope. J. Virol. 2000, 74, 7694–7697. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Harrod, K.; Shieh, W.-J.; Zaki, S.; Tripp, R.A. Human Metapneumovirus Persists in BALB/c Mice despite the Presence of Neutralizing Antibodies. J. Virol. 2004, 78, 14003–14011. [Google Scholar] [CrossRef]
- Alvarez, R.; Tripp, R.A. The Immune Response to Human Metapneumovirus Is Associated with Aberrant Immunity and Impaired Virus Clearance in BALB/c Mice. J. Virol. 2005, 79, 5971–5978. [Google Scholar] [CrossRef]
- Tzannou, I.; Nicholas, S.K.; Lulla, P.; Aguayo-Hiraldo, P.I.; Misra, A.; Martinez, C.A.; Machado, A.; Orange, J.S.; Piedra, P.A.; Vera, J.F.; et al. Immunologic Profiling of Human Metapneumovirus for the Development of Targeted Immunotherapy. J. Infect. Dis. 2017, 216, 678–687. [Google Scholar] [CrossRef]
- Wang, P.Z.; Huang, C.X.; Zhang, Y.; Li, Z.D.; Yo, H.T.; Zhang, Y.; Jia, Z.S.; Wang, J.P.; Lian, J.Q.; Sun, Y.T.; et al. Analysis of the immune response to Hantaan virus nucleocapsid protein C-terminal-specific CD8(+) T cells in patients with hemorrhagic fever with renal syndrome. Viral Immunol. 2009, 22, 253–260. [Google Scholar] [CrossRef]
- Kaukinen, P.; Vaheri, A.; Plyusnin, A. Hantavirus nucleocapsid protein: A multifunctional molecule with both housekeeping and ambassadorial duties. Arch. Virol. 2005, 150, 1693–1713. [Google Scholar] [CrossRef]
- Van Epps, H.L.; Schmaljohn, C.S.; Ennis, F.A. Human memory cytotoxic T-lymphocyte (CTL) responses to Hantaan virus infection: Identification of virus-specific and cross-reactive CD8(+) CTL epitopes on nucleocapsid protein. J. Virol. 1999, 73, 5301–5308. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Yuan, B.; Wang, M.; Zhang, Y.; Xu, Z.; Zhang, C.; Zhang, Y.; Liu, B.; Yi, J.; et al. HLA-A2 and B35 Restricted Hantaan Virus Nucleoprotein CD8+ T-Cell Epitope-Specific Immune Response Correlates with Milder Disease in Hemorrhagic Fever with Renal Syndrome. PLoS Negl. Trop. Dis. 2013, 7, e2076. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, Y.; Wang, J.; Lv, T.; Jin, B. Identification of three novel CTL epitopes within nucleocapsid protein of Hantaan virus. Viral. Immunol. 2011, 24, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Chun, E.; Kim, N.Y.; Seong, B.L. Characterization of HLA-A2.1-restricted epitopes, conserved in both Hantaan and Sin Nombre viruses, in Hantaan virus-infected patients. J. Gen. Virol. 2002, 83, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, S.; Sullivan, B.M.; Hartnett, J.N.; Robles-Sikisaka, R.; Gangavarapu, K.; Cubitt, B.; Ware, B.C.; Kotliar, D.; Branco, L.M.; Goba, A.; et al. Analysis of CD8+ T cell response during the 2013-2016 Ebola epidemic in West Africa. Proc. Natl. Acad. Sci. USA 2018, 115, E7578–E7586. [Google Scholar] [CrossRef] [PubMed]
- Sundar, K.; Boesen, A.; Coico, R. Computational prediction and identification of HLA-A2.1-specific Ebola virus CTL epitopes. Virology 2007, 360, 257–263. [Google Scholar] [CrossRef]
- ter Meulen, J.; Badusche, M.; Satoguina, J.; Strecker, T.; Lenz, O.; Loeliger, C.; Sakho, M.; Koulemou, K.; Koivogui, L.; Hoerauf, A. Old and New world arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by Lassa virus-specific human CD4+ T-cell clones. Virology 2004, 321, 134–143. [Google Scholar] [CrossRef]
- Tomori, O.; Johnson, K.M.; Kiley, M.P.; Elliot, L.H. Standardization of a plaque assay for Lassa virus. J. Med. Virol. 1987, 22, 77–89. [Google Scholar] [CrossRef]
- McElroy, A.K.; Akondy, R.S.; Harmon, J.R.; Ellebedy, A.H.; Cannon, D.; Klena, J.D.; Sidney, J.; Sette, A.; Mehta, A.K.; Kraft, C.S.; et al. A case of human Lassa virus infection with robust acute T-cell activation and long-term virus-specific T-cell responses. J. Infect. Dis. 2017, 215, 1862–1872. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Frame, J.D.; Rhoderick, J.B.; Monson, M.H. Endemic Lassa fever in Liberia: IV. Selection of optimally effective plasma for treatment by passive immunization. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 380–384. [Google Scholar] [CrossRef]
- ter Meulen, J.; Badusche, M.; Kuhnt, K.; Doetze, A.; Satoguina, J.; Marti, T.; Loeliger, C.; Koulemou, K.; Koivogui, L.; Schmitz, H.; et al. Characterization of human CD4+ T cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar] [CrossRef]
- Sakabe, S.; Hartnett, J.N.; Ngo, N.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Cubitt, B.; Garcia, S.D.; Ware, B.C.; et al. Identification of Common CD8+ T Cell Epitopes from Lassa Fever Survivors in Nigeria and Sierra Leone. J. Virol. 2020, 94, e00153-20. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Sakabe, S.; Hartnett, J.N.; Ngo, N.; Goba, A.; Momoh, M.; Sandi, J.D.; Kanneh, L.; Cubitt, B.; Garcia, S.D.; et al. High crossreactivity of human T cell responses between Lassa virus lineages. PLoS Pathog. 2020, 16, e1008352. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Lapuente, D. T Cell Immunity against Influenza: The Long Way from Animal Models Towards a Real-Life Universal Flu Vaccine. Viruses 2021, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Mao, H.; Zheng, J.; Liu, Y.; Chiu, S.S.; Qin, G.; Chan, P.L.; Lam, K.T.; Guan, J.; Zhang, L.; et al. Cytotoxic T Lymphocytes Established by Seasonal Human Influenza Cross-React against 2009 Pandemic H1N1 Influenza Virus. J. Virol. 2010, 84, 6527–6535. [Google Scholar] [CrossRef]
- Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Kreijtz, J.H.; de Mutsert, G.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Human T-cells directed to seasonal influenza A virus cross-react with 2009 pandemic influenza A (H1N1) and swine-origin triple-reassortant H3N2 influenza viruses. J. Gen. Virol. 2013, 94, 583–592. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; de Mutsert, G.; van Baalen, C.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to humaninfluenza A virus. J. Virol. 2008, 82, 5161–5166. [Google Scholar] [CrossRef]
- Lee, L.Y.H.; Ha, D.L.A.; Simmons, C.; de Jong, M.D.; Van Vinh Chau, N.; Schumacher, R.; Peng, Y.C.; McMichael, A.J.; Farrar, J.J.; Smith, G.L.; et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J. Clin. Investig. 2008, 118, 3478–3490. [Google Scholar] [CrossRef]
- Quinones-Parra, S.; Grant, E.; Loh, L.; Nguyen, T.H.; Campbell, K.A.; Tong, S.Y.; Miller, A.; Doherty, P.C.; Vijaykrishna, D.; Rossjohn, J.; et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc. Natl. Acad. Sci. USA 2014, 111, 1049–1054. [Google Scholar] [CrossRef]
- Wang, Z.; Wan, Y.; Qiu, C.; Quinones-Parra, S.; Zhu, Z.; Loh, L.; Tian, D.; Ren, Y.; Hu, Y.; Zhang, X.; et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nat. Commun. 2015, 6, 6833. [Google Scholar] [CrossRef]
- Grant, E.J.; Quiñones-Parra, S.M.; Clemens, E.B.; Kedzierska, K. Human influenza viruses and CD8(+) T cell responses. Curr. Opin. Virol. 2016, 16, 132–142. [Google Scholar] [CrossRef]
- Assarsson, E.; Bui, H.H.; Sidney, J.; Zhang, Q.; Glenn, J.; Oseroff, C.; Mbawuike, I.N.; Alexander, J.; Newman, M.J.; Grey, H.; et al. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J. Virol. 2008, 82, 12241–12251. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zanker, D.; Valkenburg, S.; Tan, B.; Kedzierska, K.; Zou, Q.M.; Doherty, P.C.; Chen, W. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 9178–9183. [Google Scholar] [CrossRef] [PubMed]
- Grant, E.; Wu, C.; Chan, K.F.; Eckle, S.; Bharadwaj, M.; Zou, Q.M.; Kedzierska, K.; Chen, W. Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol. Cell. Biol. 2013, 91, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.R.; Rothbard, J.; Gotch, F.M.; Bahadur, G.; Wraith, D.; McMichael, A.J. The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 1986, 44, 959–968. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, J.O.; Kim, J.Y.; Jung, H.E.; Lee, H.K.; Chang, J. Single mucosal vaccination targeting nucleoprotein provides broad protection against two lineages of influenza B virus. Antivir. Res. 2019, 163, 19–28. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kang, J.O.; Chang, J. Nucleoprotein vaccine induces cross-protective cytotoxic T lymphocytes against both lineages of influenza B virus. Clin. Exp. Vaccine Res. 2019, 8, 54–63. [Google Scholar] [CrossRef]
- Boon, A.C.; de Mutsert, G.; Graus, W.M.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. Sequence variation in a newly identified HLA-B35-restricted epitope in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. J. Virol. 2002, 76, 2567–2572. [Google Scholar] [CrossRef]
- Rimmelzwaan, G.F.; Boon, A.C.; Voeten, J.T.; Berkhoff, E.G.; Fouchier, R.A.; Osterhaus, A.D. Sequence variation in the influenza A virus nucleoprotein associated with escape from cytotoxic T lymphocytes. Virus Res. 2004, 103, 97–100. [Google Scholar] [CrossRef]
- Voeten, J.T.; Bestebroer, T.M.; Nieuwkoop, N.J.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J. Virol. 2000, 74, 6800–6807. [Google Scholar] [CrossRef]
- Wilkinson, T.M.; Li, C.K.F.; Chui, C.S.C.; Huang, A.K.Y.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef]
- Chen, L.; Zanker, D.; Xiao, K.; Wu, C.; Zou, Q.; Chen, W. Immunodominant CD4+ T-cell responses to influenza A virus in healthy individuals focus on matrix 1 and nucleoprotein. J. Virol. 2014, 88, 11760–11773. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, C.S.; Terry, F.E.; Peng, L.; Meza, K.A.; Sakala, I.G.; Van Aartsen, D.; Moise, L.; Martin, W.D.; Schriewer, J.; Buller, R.M.; et al. Highly conserved influenza T cell epitopes induce broadly protective immunity. Vaccine 2019, 37, 5371–5381. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.E.; Kelso, A. Prospects for an influenza vaccine that induces cross-protective cytotoxic T lymphocytes. Immunol. Cell. Biol. 2009, 87, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Liu, F.; Dong, L.; Lu, X.; Hancock, K.; Reinherz, E.L.; Katz, J.M.; Sambhara, S. Significant impact of sequence variations in the nucleoprotein on CD8 T cell-mediated cross-protection against influenza A virus infections. PLoS ONE 2010, 5, e10583. [Google Scholar] [CrossRef]
- Kreijtz, J.H.; Fouchier, R.A.; Rimmelzwaan, G.F. Immune responses to influenza virus infection. Virus Res. 2011, 162, 19–30. [Google Scholar] [CrossRef]
- Hayward, A.C.; Wang, L.; Goonetilleke, N.; Fragaszy, E.B.; Bermingham, A.; Copas, A.; Dukes, O.; Millett, E.R.; Nazareth, I.; Nguyen-Van-Tam, J.S.; et al. Natural t cell-mediated protection against seasonal and pandemic influenza. Results of the flu watch cohort study. Am. J. Respir. Crit. Care Med. 2015, 191, 1422–1431. [Google Scholar] [CrossRef]
- Boon, A.C.; de Mutsert, G.; van Baarle, D.; Smith, D.J.; Lapedes, A.S.; Fouchier, R.A.; Sintnicolaas, K.; Osterhaus, A.D.; Rimmelzwaan, G.F. Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J. Immunol. 2004, 172, 2453–2460. [Google Scholar] [CrossRef]
- Gschoesser, C.; Almanzar, G.; Hainz, U.; Ortin, J.; Schonitzer, D.; Schild, H.; Saurwein-Teissl, M.; Grubeck-Loebenstein, B. CD4+ and CD8+ mediated cellular immune response to recombinant influenza nucleoprotein. Vaccine 2002, 20, 3731–3738. [Google Scholar] [CrossRef]
- Brett, S.J.; Blau, J.; Hughes-Jenkins, C.M.; Rhodes, J.; Liew, F.Y.; Tite, J.P. Human T cell recognition of influenza A nucleoprotein. Specificity and genetic restriction of immunodominant T helper cell epitopes. J. Immunol. 1991, 147, 984–991. [Google Scholar]
- Townsend, A.; Bastin, J.; Bodmer, H.; Brownlee, G.; Davey, J.; Gotch, F.; Gould, K.; Jones, I.; McMichael, A.; Rothbard, J. Recognition of influenza virus proteins by cytotoxic T lymphocytes. Philos. Trans. R. Soc. B Biol. Sci. 1989, 323, 527–533. [Google Scholar] [CrossRef]
- McMichael, A.J.; Michie, C.A.; Gotch, F.M.; Smith, G.L.; Moss, B. Recognition of influenza A virus nucleoprotein by human cytotoxic T lymphocytes. J. Gen. Virol. 1986, 67, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Bennink, J.R.; Smith, G.L.; Moss, B. Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 1785–1789. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Q.M.; Gatherer, D.; Reche, P.A.; Flower, D.R. Towards the knowledge-based design of universal influenza epitope ensemble vaccines. Bioinformatics 2016, 32, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.S.; Ke, F.; Grigorova, I.L. B Cell Receptor Crosslinking Augments Germinal Center B Cell Selection when T Cell Help Is Limiting. Cell Rep. 2018, 25, 1395–1403. [Google Scholar] [CrossRef]
- Matić, Z.; Šantak, M. Current view on novel vaccine technologies to combat human infectious diseases. Appl. Microbiol. Biotechnol. 2021, 10, 1–32. [Google Scholar] [CrossRef]
- Carragher, D.M.; Kaminski, D.A.; Moquin, A.; Hartson, L.; Randall, T.D. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J. Immunol. 2008, 181, 4168–4176. [Google Scholar] [CrossRef]
- LaMere, M.W.; Lam, H.T.; Moquin, A.; Haynes, L.; Lund, F.E.; Randall, T.D.; Kaminski, D.A. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J. Immunol. 2011, 186, 4331–4339. [Google Scholar] [CrossRef]
- Díaz, F.E.; Guerra-Maupome, M.; McDonald, P.O.; Rivera-Pérez, D.; Kalergis, A.M.; McGill, J.L. A Recombinant BCG Vaccine Is Safe and Immunogenic in Neonatal Calves and Reduces the Clinical Disease Caused by the Respiratory Syncytial Virus. Front. Immunol. 2021, 12, 664212. [Google Scholar] [CrossRef]
- Roux, X.; Dubuquoy, C.; Durand, G.; Tran-Tolla, T.L.; Castagné, N.; Bernard, J.; Petit-Camurdan, A.; Eléouët, J.F.; Riffault, S. Sub-nucleocapsid nanoparticles: A nasal vaccine against respiratory syncytial virus. PLoS ONE 2008, 3, e1766. [Google Scholar] [CrossRef]
- Remot, A.; Roux, X.; Dubuquoy, C.; Fix, J.; Bouet, S.; Moudjou, M.; Eléouët, J.F.; Riffault, S.; Petit-Camurdan, A. Nucleoprotein nanostructures combined with adjuvants adapted to the neonatal immune context: A candidate mucosal RSV vaccine. PLoS ONE 2012, 7, e37722. [Google Scholar] [CrossRef]
- Gott, P.; Zoller, L.; Darai, G.; Bautz, E.K. A major antigenic domain of hantaviruses is located on the aminoproximal site of the viral nucleocapsid protein. Virus Genes 1997, 14, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kallio-Kokko, H.; Lundkvist, A.; Plyusnin, A.; Avsic-Zupanc, T.; Vaheri, A.; Vapalahti, O. Antigenic properties and diagnostic potential of recombinant Dobrava virus nucleocapsid protein. J. Med. Virol. 2000, 61, 266–274. [Google Scholar] [CrossRef]
- Kallio-Kokko, H.; Leveelahti, R.; Brummer-Korvenkontio, M.; Lundkvist, A.; Vaheri, A.; Vapalahti, O. Human immune response to Puumala virus glycoproteins and nucleocapsid protein expressed in mammalian cells. J. Med. Virol. 2001, 65, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, A.; Bjorsten, S.; Niklasson, B.; Ahlborg, N. Mapping of B-cell determinants in the nucleocapsid protein of Puumala virus: Definition of epitopes specific for acute immunoglobulin G recognition in humans. Clin. Diagn. Lab. Immunol. 1995, 2, 82–86. [Google Scholar] [CrossRef]
- Lundkvist, A.; Kallio-Kokko, H.; Sjolander, K.B.; Lankinen, H.; Niklasson, B.; Vaheri, A.; Vapalahti, O. Characterization of Puumala virus nucleocapsid protein: Identification of B-cell epitopes and domains involved in protective immunity. Virology 1996, 216, 397–406. [Google Scholar] [CrossRef]
- Lundkvist, A.; Vapalahti, O.; Plyusnin, A.; Sjolander, K.B.; Niklasson, B.; Vaheri, A. Characterization of Tula virus antigenic determinants defined by monoclonal antibodies raised against baculovirus-expressed nucleocapsid protein. Virus Res. 1996, 45, 29–44. [Google Scholar] [CrossRef]
- Tischler, N.D.; Galeno, H.; Rosemblatt, M.; Valenzuela, P.D. Human and rodent humoral immune responses to Andes virus structural proteins. Virology 2005, 334, 319–326. [Google Scholar] [CrossRef]
- Zoller, L.; Scholz, J.; Stohwasser, R.; Giebel, L.B.; Sethi, K.K.; Bautz, E.K.; Darai, G. Immunoblot analysis of the serological response in Hantavirus infections. J. Med. Virol. 1989, 27, 231–237. [Google Scholar] [CrossRef]
- Maes, P.; Clement, J.; Gavrilovskaya, I.; Van Ranst, M. Hantaviruses: Immunology, treatment and prevention. Viral Immunol. 2004, 17, 481–497. [Google Scholar] [CrossRef]
- Li, Y.; Quan, C.; Xing, W.; Wang, P.; Gao, J.; Zhang, Z.; Jiang, X.; Ma, C.; Carr, M.J.; He, Q.; et al. Rapid humoral immune responses are required for recovery from haemorrhagic fever with renal syndrome patients. Emerg. Microbes Infect. 2020, 9, 2303–2314. [Google Scholar] [CrossRef]
- Fernie, B.F.; Ford, E.C.; Gerin, J.L. The development of Balb/c cells persistently infected with respiratory syncytial virus: Presence of ribonucleoprotein on the cell surface. Proc. Soc. Exp. Biol. Med. 1981, 167, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Zeller, W.; Bruns, M.; Lehmann-Grube, F. Lymphocytic choriomeningitis virus. X. Demonstration of nucleoprotein on the surface of infected cells. Virology 1988, 162, 90–97. [Google Scholar] [CrossRef]
- Virelizier, J.L.; Allison, A.C.; Oxford, J.S.; Schild, G.C. Early presence of ribonucleoprotein antigen on surface of influenza virus-infected cells. Nature 1977, 266, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Frank, E.; Gerhard, W. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J. Immunol. 1981, 126, 1814–1819. [Google Scholar]
- Prokudina, E.N.; Semenova, N.P.; Yamnikova, S.S.; Zhdanov, V.M. Effect of β-decay of radionuclides incorporated into influenza virus RNA and proteins on the infectivity of the virus and antigenicity of its nucleoprotein. Arch. Virol. 1987, 97, 326–332. [Google Scholar] [CrossRef]
- Prokudina, E.N.; Semenova, N.P. Localization of the influenza virus nucleoprotein: Cell-associated and extracellular non-virion forms. J. Gen. Virol. 1991, 72, 1699–1702. [Google Scholar] [CrossRef]
- Bodewes, R.; Geelhoed-Mieras, M.M.; Wrammert, J. In vitro assessment of the immunological significance of a human monoclonal antibody directed to the influenza a virus nucleoprotein. Clin. Vaccine Immunol. 2013, 20, 1333–1337. [Google Scholar] [CrossRef]
- Gui, X.; Ge, P.; Wang, X.; Yang, K.; Yu, H.; Zhao, Q.; Chen, Y.; Xia, N. Identification of a highly conserved and surface exposed B-cell epitope on the nucleoprotein of influenza A virus. J. Med. Virol. 2014, 86, 995–1002. [Google Scholar] [CrossRef]
- Vanderven, H.A.; Ana-Sosa-Batiz, F.; Jegaskanda, S.; Rockman, S.; Laurie, K.; Barr, I.; Chen, W.; Wines, B.; Hogarth, P.M.; Lambe, T.; et al. What Lies Beneath: Antibody Dependent Natural Killer Cell Activation by Antibodies to Internal Influenza Virus Proteins. EBioMedicine 2016, 8, 277–290. [Google Scholar] [CrossRef]
- McEwan, W.A.; Tam, J.C.; Watkinson, R.E.; Bidgood, S.R.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, D.; Zhao, B.; Cao, Y.; Yu, J.; Yan, H.; Zhao, W.; Zhang, E.; Yang, J.; Zhong, M.; et al. Immunoglobulin A Targeting on the N-Terminal Moiety of Viral Phosphoprotein Prevents Measles Virus from Evading Interferon-β Signaling. ACS Infect. Dis. 2020, 6, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Yang, Y.; Zhao, B.; Yu, J.; Cao, Y.; Yan, H.; Zhao, W.; Chen, L.; Chen, F.; Li, X.; et al. IgA targeting on the α-molecular recognition element (α-MoRE) of viral phosphoprotein inhibits measles virus replication by interrupting formation and function of P-N complex intracellularly. Antivir. Res. 2019, 161, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, Y.; Li, Q.; Chen, Y.; He, B.; Yang, J.; Tu, H.; Lei, L.; Yan, H. Matrix Protein-Specific IgA Antibody Inhibits Measles Virus Replication by Intracellular Neutralization. J. Virol. 2011, 85, 11090–11097. [Google Scholar] [CrossRef] [PubMed]
- Lepault, J.; Petitpas, I.; Erk, I.; Navaza, J.; Bigot, D.; Dona, M.; Vachette, P.; Cohen, J.; Rey, F.A. Structural polymorphism of the major capsid protein of rotavirus. EMBO J. 2001, 20, 1498–1507. [Google Scholar] [CrossRef]
- Bugli, F.; Caprettini, V.; Cacaci, M.; Martini, C.; Paroni Sterbini, F.; Torelli, R.; Della Longa, S.; Papi, M.; Palmieri, V.; Giardina, B.; et al. Synthesis and characterization of different immunogenic viral nanoconstructs from rotavirus VP6 inner capsid protein. Int. J. Nanomed. 2014, 9, 2727–2739. [Google Scholar]
- Thouvenin, E.; Schoehn, G.; Rey, F.; Petitpas, I.; Mathieu, M.; Vaney, M.C.; Cohen, J.; Kohli, E.; Pothier, P.; Hewat, E. Antibody inhibition of the transcriptase activity of the rotavirus DLP: A structural view. J. Mol. Biol. 2001, 307, 161–172. [Google Scholar] [CrossRef]
- Aiyegbo, M.S.; Sapparapu, G.; Spiller, B.W.; Eli, I.M.; Williams, D.R.; Kim, R.; Lee, D.E.; Liu, T.; Li, S.; Woods, V.L., Jr.; et al. Human rotavirus VP6-specific antibodies mediate intracellular neutralization by binding to a quaternary structure in the transcriptional pore. PLoS ONE 2013, 8, e61101. [Google Scholar] [CrossRef]
- Bai, Y.; Ye, L.; Tesar, D.B.; Song, H.; Zhao, D.; Björkman, P.J.; Roopenian, D.C.; Zhu, X. Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proc. Natl. Acad. Sci. USA 2011, 108, 18406–18411. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Straub, T.; Schweier, O.; Bruns, M.; Nimmerjahn, F.; Waisman, A.; Pircher, H. Nucleoprotein-specific nonneutralizing antibodies speed up LCMV elimination independently of complement and FcγR. Eur. J. Immunol. 2013, 43, 2338–2348. [Google Scholar] [CrossRef]
- Joseph, B.S.; Oldstone, M.B. Antibody-induced redistribution of measles virus antigens on the cell surface. J. Immunol. 1974, 113, 1205–1209. [Google Scholar] [PubMed]
- Kajihara, M.; Marzi, A.; Nakayama, E.; Noda, T.; Kuroda, M.; Manzoor, R.; Matsuno, K.; Feldmann, H.; Yoshida, R.; Kawaoka, Y.; et al. Inhibition of Marburg virus budding by nonneutralizing antibodies to the envelope glycoprotein. J. Virol. 2012, 86, 13467–13474. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.W.; Portner, A.; Murti, K.G. Antibodies to paramyxovirus nucleoproteins define regions important for immunogenicity and nucleocapsid assembly. Virology 1993, 193, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP + M1. Clin. Infect. Dis. 2011, 52, 1–7. [Google Scholar] [CrossRef]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.; et al. A T cell-inducing influenza vaccine for the elderly: Safety and immunogenicity of MVA-NP + M1 in adults aged over 50 years. PLoS ONE 2012, 7, e48322. [Google Scholar] [CrossRef]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.A.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP + M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef]
- Swayze, H.; Allen, J.; Folegatti, P.; Yu, L.M.; Gilbert, S.; Hill, A.; Ellis, C.; Butler, C.C. A phase IIb study to determine the safety and efficacy of candidate INfluenza Vaccine MVA-NP + M1 in combination with licensed Ina CTivated infl Uenza vaccine in adult S aged 65 years and above (INVICTUS): A study protocol. F1000Research 2019, 8, 719. [Google Scholar] [CrossRef]
- Butler, C.; Ellis, C.; Folegatti, P.M.; Swayze, H.; Allen, J.; Bussey, L.; Bellamy, D.; Lawrie, A.; Eagling-Vose, E.; Yu, L.M.; et al. Efficacy and Safety of a Modified Vaccinia Ankara-NP + M1 Vaccine Combined with QIV in People Aged 65 and Older: A Randomised Controlled Clinical Trial (INVICTUS). Vaccines 2021, 9, 851. [Google Scholar] [CrossRef]
- Dicks, M.D.; Spencer, A.J.; Edwards, N.J.; Wadell, G.; Bojang, K.; Gilbert, S.C.; Hill, A.V.S.; Cottingham, M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS ONE 2012, 7, e40385. [Google Scholar] [CrossRef]
- Antrobus, R.D.; Coughlan, L.; Berthoud, T.K.; Dicks, M.D.; Hill, A.V.; Lambe, T.; Gilbert, S.C. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved Influenza A antigens. Mol. Ther. 2014, 22, 668–674. [Google Scholar] [CrossRef]
- Del Campo, J.; Pizzorno, A.; Djebali, S.; Bouley, J.; Haller, M.; Pérez-Vargas, J.; Lina, B.; Boivin, G.; Hamelin, M.E.; Nicolas, F.; et al. OVX836 a recombinant nucleoprotein vaccine inducing cellular responses and protective efficacy against multiple influenza A subtypes. NPJ Vaccines 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Meisinger-Henschel, C.; Tzatzaris, M.; Hülsemann, V.; Lukassen, S.; Wulff, N.H.; Hausmann, J.; Howley, P.; Chaplin, P. Modified vaccinia Ankara strains with identical coding sequences actually represent complex mixtures of viruses that determine the biological properties of each strain. Vaccine 2009, 27, 7442–7450. [Google Scholar] [CrossRef] [PubMed]
- Samy, N.; Reichhardt, D.; Schmidt, D.; Chen, L.M.; Silbernagl, G.; Vidojkovic, S.; Meyer, T.P.; Jordan, E.; Adams, T.; Weidenthaler, H.; et al. Safety and immunogenicity of novel modified vaccinia Ankara-vectored RSV vaccine: A randomized phase I clinical trial. Vaccine 2020, 38, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Lawrence, S.J.; Meyer, T.P.H.; Schmidt, D.; Schultz, S.; Mueller, J.; Stroukova, D.; Koenen, B.; Gruenert, R.; Silbernagl, G.; et al. Broad Antibody and Cellular Immune Response from a Phase 2 Clinical Trial with a Novel Multivalent Poxvirus-Based Respiratory Syncytial Virus Vaccine. J. Infect. Dis. 2021, 223, 1062–1072. [Google Scholar] [CrossRef]
- Cicconi, P.; Jones, C.; Sarkar, E.; Silva-Reyes, L.; Klenerman, P.; de Lara, C.; Hutchings, C.; Moris, P.; Janssens, M.; Fissette, L.A.; et al. First-in-Human Randomized Study to Assess the Safety and Immunogenicity of an Investigational Respiratory Syncytial Virus (RSV) Vaccine Based on Chimpanzee-Adenovirus-155 Viral Vector-Expressing RSV Fusion, Nucleocapsid, and Antitermination Viral Proteins in Healthy Adults. Clin. Infect. Dis. 2020, 70, 2073–2081. [Google Scholar]
- Abarca, K.; Rey-Jurado, E.; Muñoz-Durango, N.; Vázquez, Y.; Soto, J.A.; Gálvez, N.M.S.; Valdés-Ferrada, J.; Iturriaga, C.; Urzúa, M.; Borzutzky, A.; et al. Safety and immunogenicity evaluation of recombinant BCG vaccine against respiratory syncytial virus in a randomized, double-blind, placebo-controlled phase I clinical trial. EClinicalMedicine 2020, 27, 100517. [Google Scholar] [CrossRef]
- Pollard, A.J.; Launay, O.; Lelievre, J.D.; Lacabaratz, C.; Grande, S.; Goldstein, N.; Robinson, C.; Gaddah, A.; Bockstal, V.; Wiedemann, A.; et al. Safety and immunogenicity of a two-dose heterologous Ad26.ZEBOV and MVA-BN-Filo Ebola vaccine regimen in adults in Europe (EBOVAC2): A randomised, observer-blind, participant-blind, placebo-controlled, phase 2 trial. Lancet Infect. Dis. 2021, 21, 493–506. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Kikuchi, K.; Maity, B.; Ueno, T. The Versatile Manipulations of Self-Assembled Proteins in Vaccine Design. Int. J. Mol. Sci. 2012, 22, 1934. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Zottig, X.; Côté-Cyr, M.; Arpin, D.; Archambault, D.; Bourgault, S. Protein Supramolecular Structures: From Self-Assembly to Nanovaccine Design. Nanomaterials 2020, 10, 1008. [Google Scholar] [CrossRef]
- Hervé, P.L.; Raliou, M.; Bourdieu, C.; Dubuquoy, C.; Petit-Camurdan, A.; Bertho, N.; Eléouët, J.F.; Chevalier, C.; Riffault, S. A novel subnucleocapsid nanoplatform for mucosal vaccination against influenza virus that targets the ectodomain of matrix protein 2. J. Virol. 2014, 88, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.; Ruffie, C.; Dubois, M.; Combredet, C.; Amino, R.; Formaglio, P.; Gorgette, O.; Pehau-Arnaudet, G.; Guery, C.; Puijalon, O.; et al. Whole Pichia pastoris Yeast Expressing Measles Virus Nucleoprotein as a Production and Delivery System to Multimerize Plasmodium Antigens. PLoS ONE 2014, 9, e86658. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.; Ruffie, C.; Combredet, C.; Formaglio, P.; Amino, R.; Ménard, R.; Tangy, F.; Sala, M. Yeast lysates carrying the nucleoprotein from measles virus vaccine as a novel subunit vaccine platform to deliver Plasmodium circumsporozoite antigen. Malar. J. 2017, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Mebatsion, T.; Koolen, M.J.M.; de Vaan, L.T.C.; de Haas, N.; Braber, M.; Römer-Oberdörfer, A.; van den Elzen, P.; van der Marel, P. Newcastle disease virus (NDV) marker vaccine: An immunodominant epitope on the nucleoprotein gene of NDV can be deleted or replaced by a foreign epitope. J. Virol. 2002, 76, 10138–10146. [Google Scholar] [CrossRef]



| Location | Sequence | HLA Antigen 1 | Reference |
|---|---|---|---|
| 271–290 | LTIKFGIETMYPALGLHEFA | n.d. | [128] |
| 367–386 | EMVRRSAGKVSSTLASELGI | n.d. | |
| 400–420 | TTEDKISRAVGPRQAQVSFL | n.d. | |
| 483–502 | QDPQDSRRSAEPLLRLQAMA | n.d. | |
| 185–199 | PDTAADSELRRWIKY | HLA-DRB1*1103 | [130] |
| 321–340 | QNKFSAGSYPLLWSYAMGVG | n.d. | [129] |
| 331–350 | LLWSYAMGVGVELENSMGGL | n.d. | |
| 372–385 | SAGKVSSTLASELG | HLA-DRB1*0301 | [131] |
| Location | Sequence | HLA Antigen 1 | Cross-Reactivity to Distantly Related Viruses 1 | Reference |
|---|---|---|---|---|
| 12–20 | NAHEGQLVI | HLA-B51 | yes | [152] |
| 129–137 | FVVPILLKA | HLA-A2 | yes | [153] |
| 131–139 | VPILLKALY | HLA-B35 | yes | [153] |
| 167–175 | DVNGIRKPK | HLA-A33 | yes | [153] |
| 197–205 | RYRTAVCGL | HLA-A11 | yes | [154] |
| 245–253 | KLLPDTAAV | HLA-A24 | yes | [154] |
| 247–255 | LPDTAAVSL | HLA-B35 | no | [153] |
| 258–266 | GPATNRDYL | HLA-B7 | yes | [154] |
| 277–285 | ETKESKAIR | HLA-A33 | no | [153] |
| 301–315 | SPSSIWVFAGAPDRC | n.d. | n.d. | [154] |
| 334–342 | ILQDMRNTI | HLA-A2.1 | yes | [155] |
| 355–369 | LRKKSSFYQSYLRRT | n.d. | n.d. | [154] |
| 415–429 | DVKVKEISNQEPLKL | n.d. | n.d. | [154] |
| 421–429 | ISNQEPLKL | HLA-A1 | yes | [152] |
| Medical Condition | Clinical Trial Identifier (Regulatory Agency) | Vaccine Type | Phase (Status) | Sponsor |
|---|---|---|---|---|
| influenza | 2009-010334-21 (EMA) | MVA * encoding NP and M1 proteins (MVA-NP + M1) | IIa (completed in 2010) | University of Oxford |
| NCT00993083 (FDA) | II (completed in 2010) | |||
| NCT01818362 (FDA) | chimpanzee adenovirus AdOx1 encoding NP and M1 (ChAdOx1 NP + M1) | I (completed in 2015) | ||
| 2017-001103-77 (EMA) | seasonal inactivated influenza vaccine in combination with MVA-NP + M1 | IIb (completed in 2018) | Vaccitech Limited | |
| NCT03300362 (FDA) | IIb (completed in 2018) | |||
| 2021-002535-39 (EMA) | oligomerization domain OVX313 fused to NP which formed the NP heptamer (OVX836) | IIb (ongoing) | Osivax S.A.S | |
| NCT03594890 (FDA) | I (completed in 2019) | |||
| RSV | 2017-004582-27 (EMA) | MVA * encoding RSV antigens F, G (of subtypes A and B), NP and M2 (MVA-BN-RSV) | IIa (completed in 2019) | Bavarian Nordic |
| NCT04752644 (FDA) | II (ongoing) | |||
| 2018-000431-27 (EMA) | chimpanzee adenovirus Ad155 encoding F, NP and M2 proteins (ChAd155-RSV) | I/II (completed in 2021) | GlaxoSmithKline Biologicals | |
| NCT02491463 (FDA) | I (completed in 2017) | |||
| NCT03213405 (FDA) | BCG ** encoding RSV NP (rBCG-N-hRSV) | I (completed in 2018) | UC Chile | |
| Ebola | NCT04152486 | MVA * encoding glycoproteins of Zaire ebolavirus, Sudan ebolavirus and Marburg Marburgvirus, and NP of Taï Forest ebolavirus (MVA-BN-Filo) | III (ongoing) | London School of Hygiene and Tropical Medicine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šantak, M.; Matić, Z. The Role of Nucleoprotein in Immunity to Human Negative-Stranded RNA Viruses—Not Just Another Brick in the Viral Nucleocapsid. Viruses 2022, 14, 521. https://doi.org/10.3390/v14030521
Šantak M, Matić Z. The Role of Nucleoprotein in Immunity to Human Negative-Stranded RNA Viruses—Not Just Another Brick in the Viral Nucleocapsid. Viruses. 2022; 14(3):521. https://doi.org/10.3390/v14030521
Chicago/Turabian StyleŠantak, Maja, and Zrinka Matić. 2022. "The Role of Nucleoprotein in Immunity to Human Negative-Stranded RNA Viruses—Not Just Another Brick in the Viral Nucleocapsid" Viruses 14, no. 3: 521. https://doi.org/10.3390/v14030521
APA StyleŠantak, M., & Matić, Z. (2022). The Role of Nucleoprotein in Immunity to Human Negative-Stranded RNA Viruses—Not Just Another Brick in the Viral Nucleocapsid. Viruses, 14(3), 521. https://doi.org/10.3390/v14030521