
Replication Kinetics for a Reporter Merkel Cell Polyomavirus

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Plasmids and Constructs
2.3. Recircularization of MCV Genome by In Vitro Ligation and Mini-Circle System
2.4. MCVmc Transfection and Replication Assay
2.5. Quantitation of MCV Genome Copy by Real-Time PCR
2.6. Immunoblotting
2.7. Immunofluorescence Assay (IFA)
2.8. Virion Production
2.9. Electron Microscopy
2.10. MCV Infection Assay
2.11. Southern Blot
2.12. MCV Kinetics Assay
2.13. Flow Cytometry
2.14. MCV Packaging Assay
3. Results
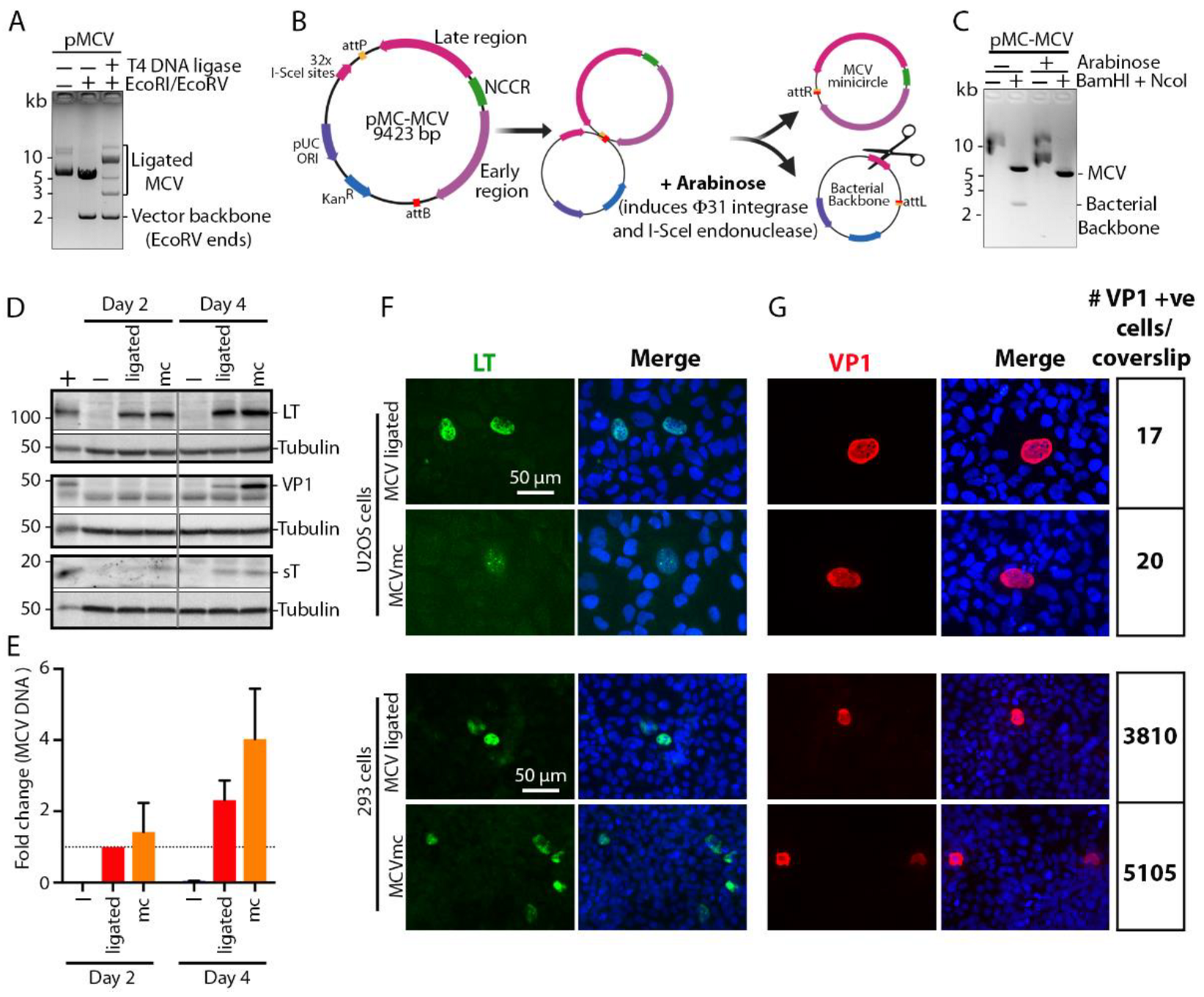
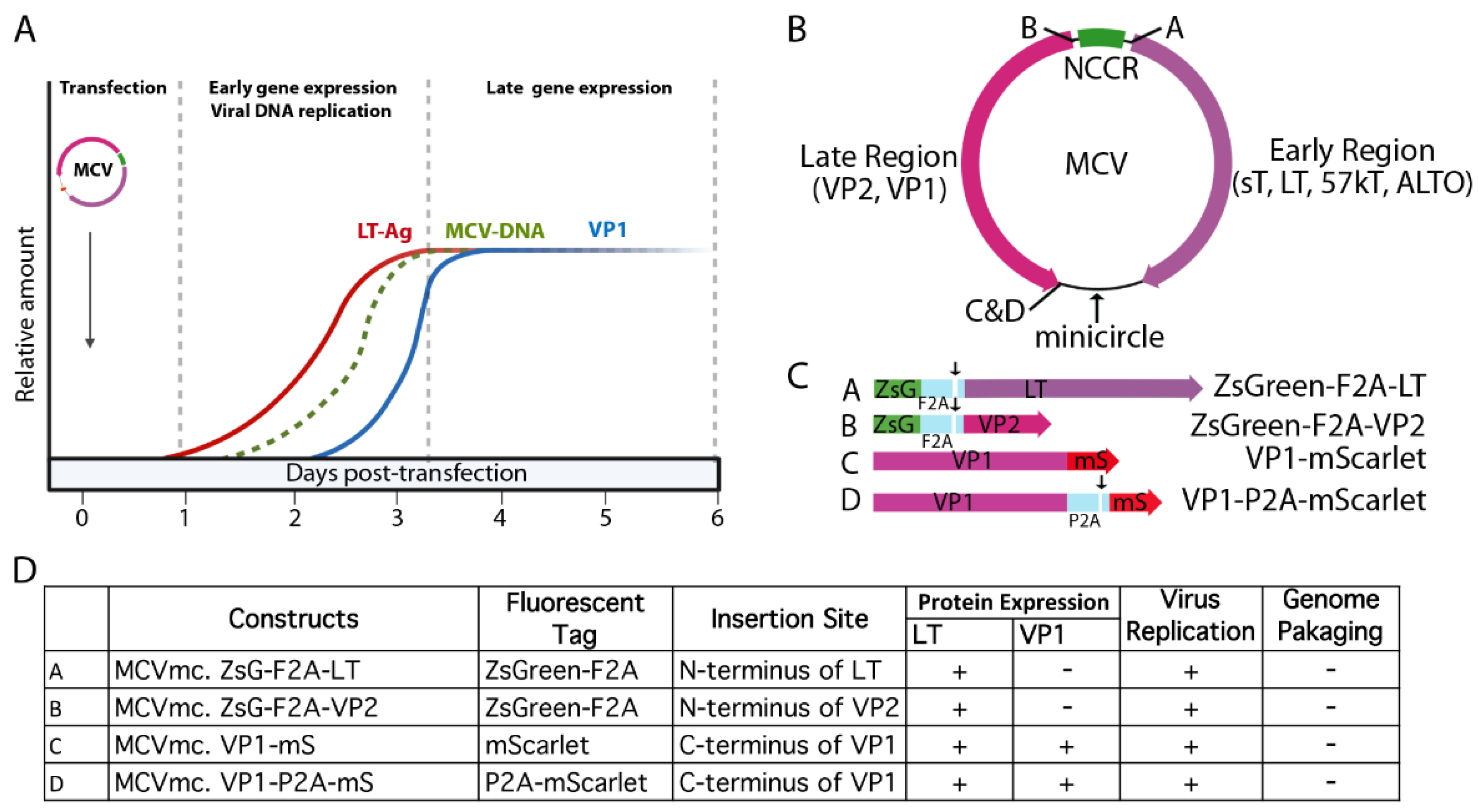
3.1. MCV Genome Recircularization by Site-Specific Recombination
3.2. MCVmc Gene Expression
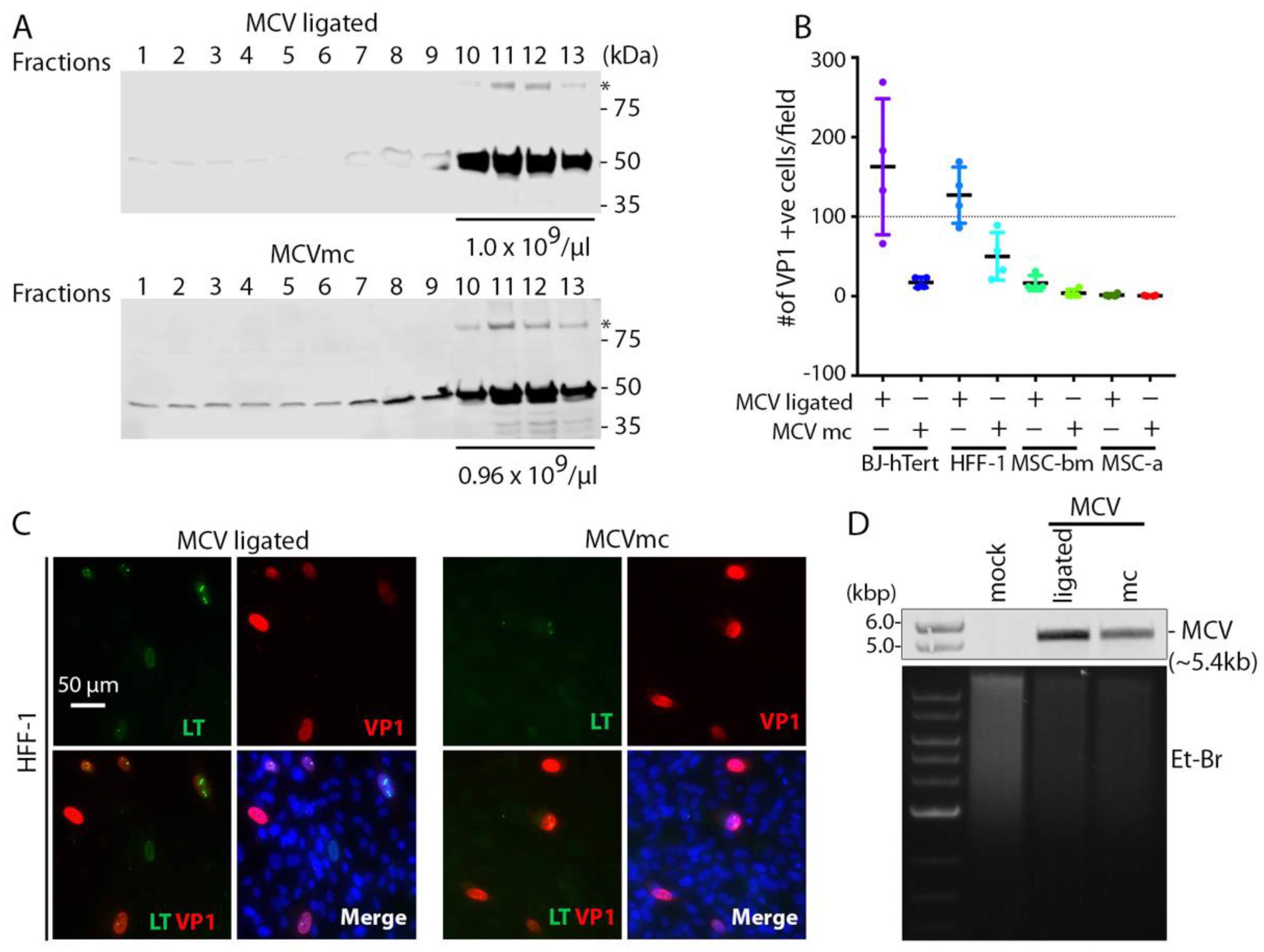
3.3. MCVmc In Vitro Transmission
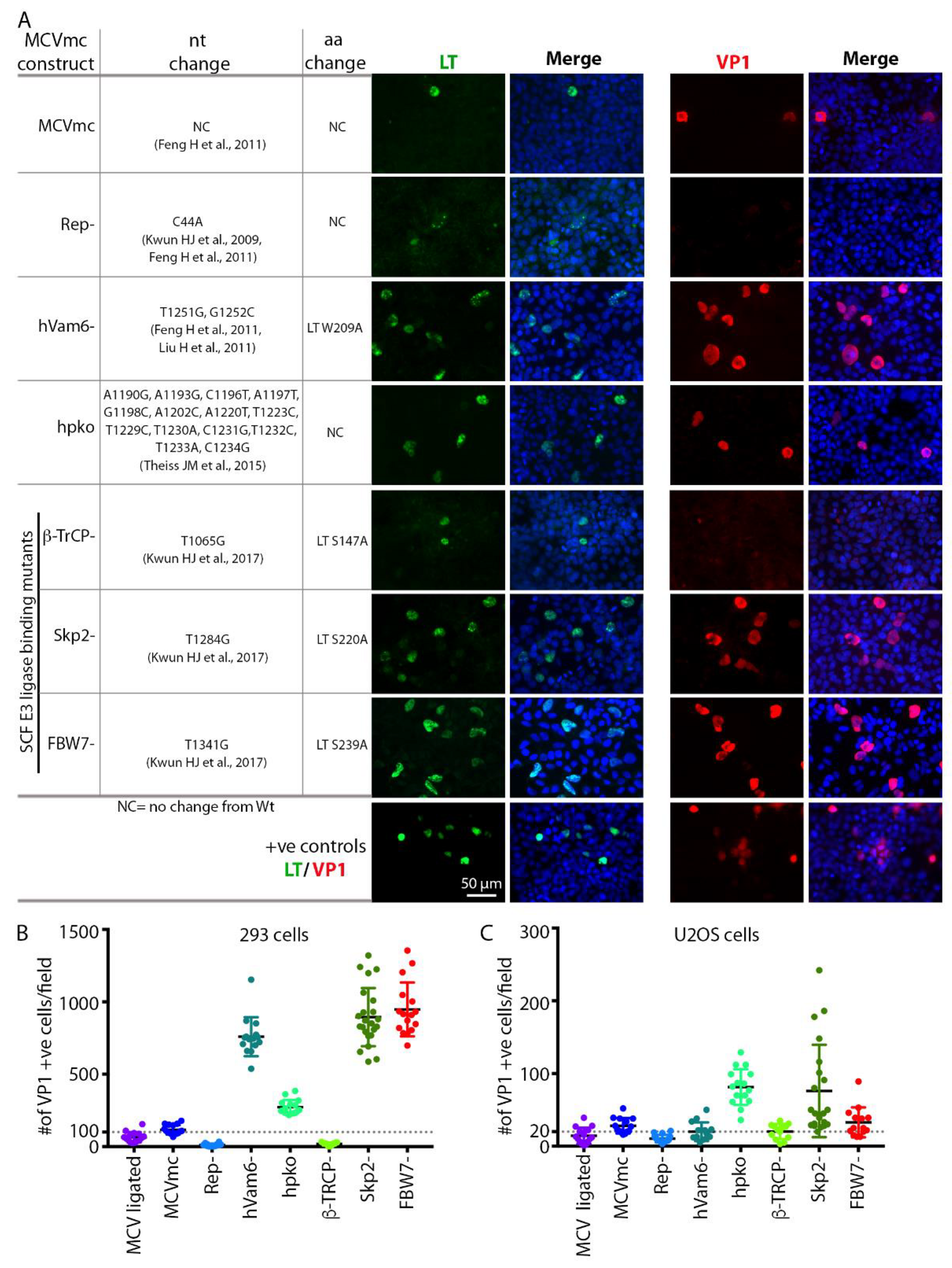
3.4. Mutagenesis of the MCVmc Genome and Cell-Specific Effects on Replication
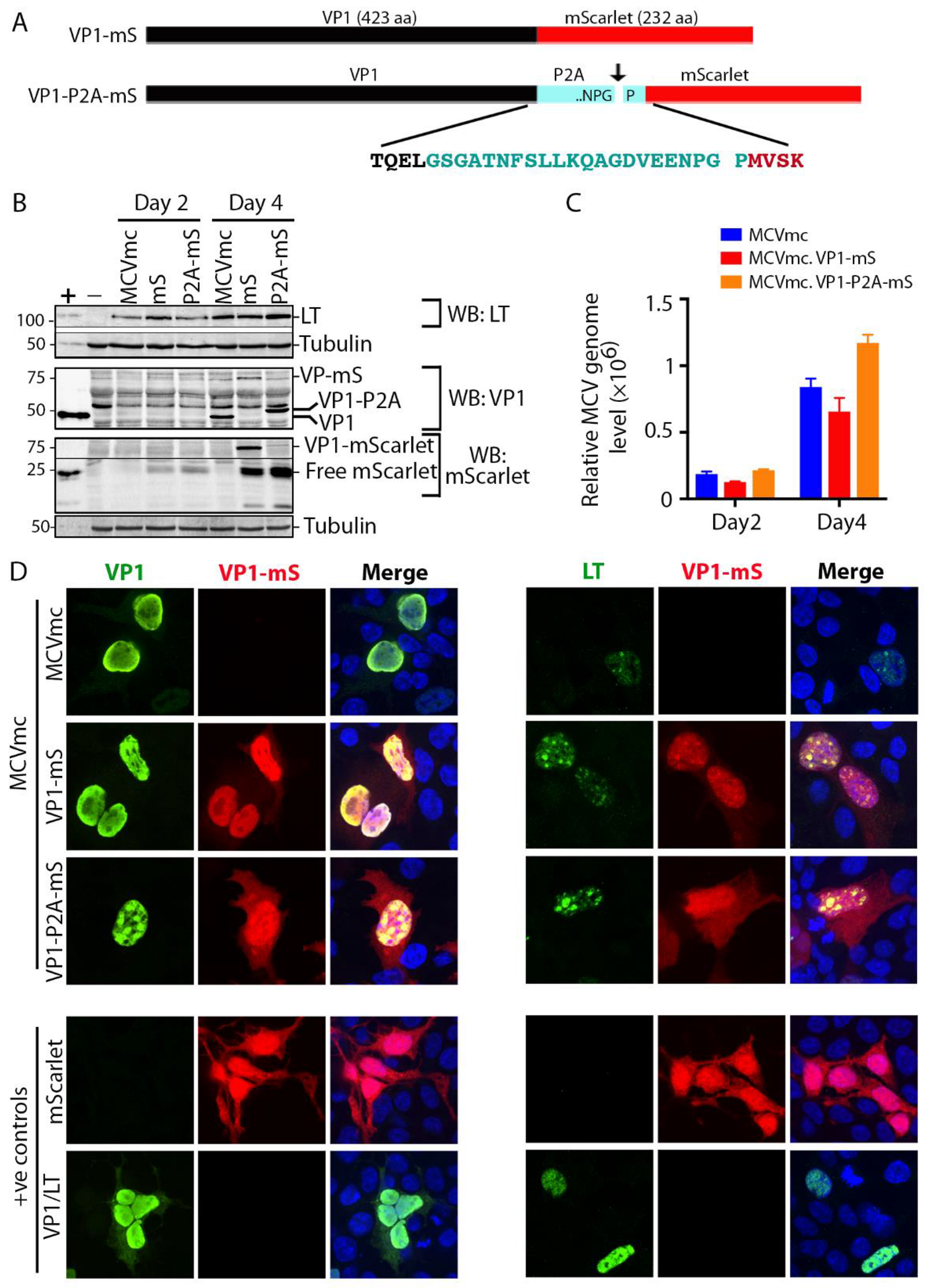
3.5. Generation of Fluorescent MCV Reporter Viruses (MCVmc.VP1-mS and MCVmc.VP1-P2A-mS)
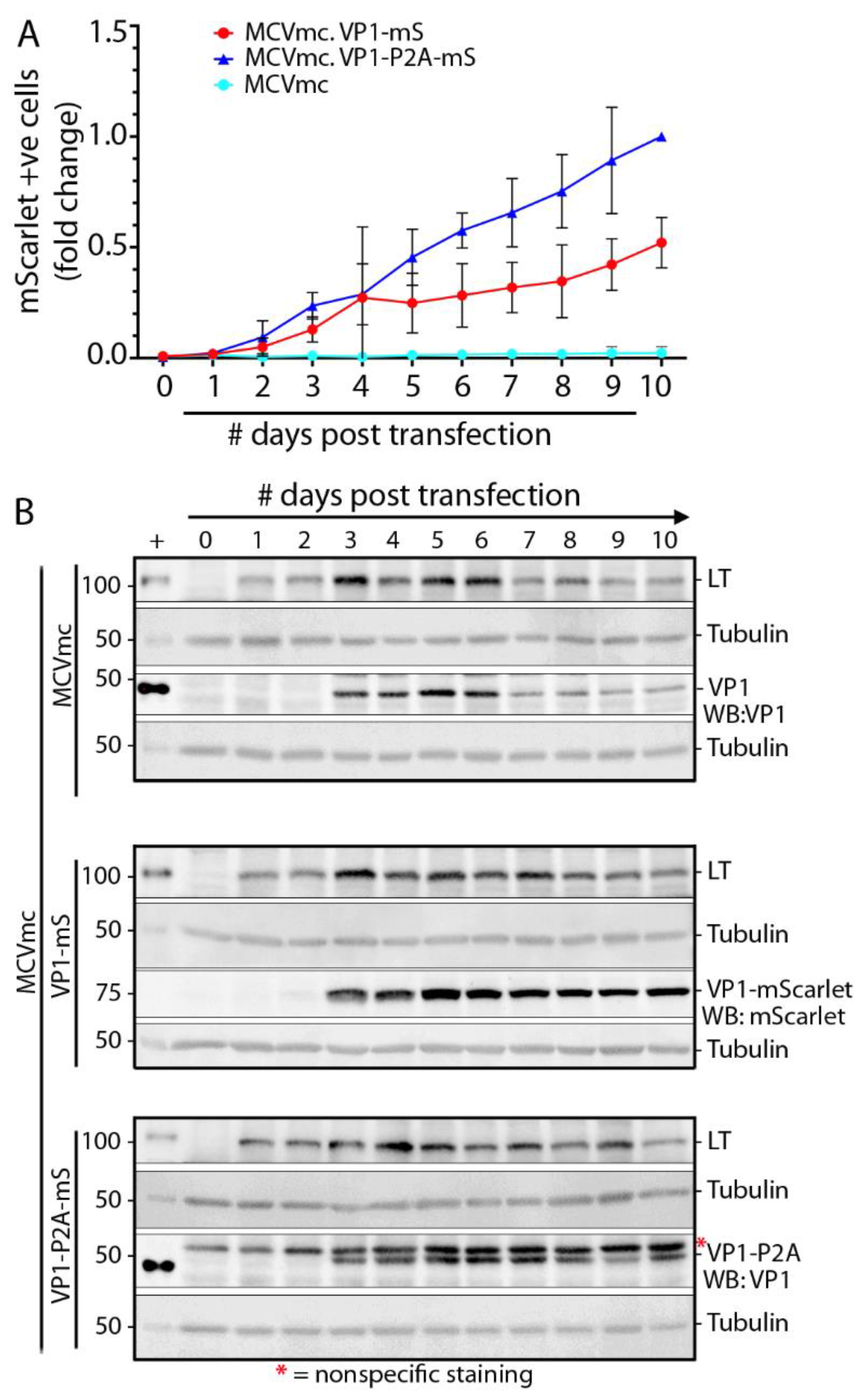
3.6. Replication Kinetics for MCV Reporter Viruses
3.7. Single-Round Transmission of MCV mScarlet Reporter Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e2. [Google Scholar] [CrossRef] [PubMed]
- Lemos, B.D.; Storer, B.E.; Iyer, J.G.; Phillips, J.L.; Bichakjian, C.K.; Fang, L.C.; Johnson, T.M.; Liegeois-Kwon, N.J.; Otley, C.C.; Paulson, K.G.; et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5823 cases as the basis of the first consensus staging system. J. Am. Acad. Dermatol. 2010, 63, 751–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, e02079-16. [Google Scholar] [CrossRef] [Green Version]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The Minimum Replication Origin of Merkel Cell Polyomavirus Has a Unique Large T-Antigen Loading Architecture and Requires Small T-Antigen Expression for Optimal Replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Theiss, J.M.; Günther, T.; Alawi, M.; Neumann, F.; Tessmer, U.; Fischer, N.; Grundhoff, A. A Comprehensive Analysis of Replicating Merkel Cell Polyomavirus Genomes Delineates the Viral Transcription Program and Suggests a Role for mcv-miR-M1 in Episomal Persistence. PLoS Pathog. 2015, 11, e1004974. [Google Scholar] [CrossRef]
- Abere, B.; Zhou, H.; Li, J.; Cao, S.; Toptan, T.; Grundhoff, A.; Fischer, N.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Encodes Circular RNAs (circRNAs) Enabling a Dynamic circRNA/microRNA/mRNA Regulatory Network. mBio 2020, 11, e03059-20. [Google Scholar] [CrossRef]
- Yang, R.; Lee, E.E.; Kim, J.; Choi, J.H.; Kolitz, E.; Chen, Y.; Crewe, C.; Salisbury, N.J.; Scherer, P.E.; Cockerell, C.; et al. Characterization of ALTO-encoding circular RNAs expressed by Merkel cell polyomavirus and trichodysplasia spinulosa polyomavirus. PLoS Pathog. 2021, 17, e1009582. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel Cell Polyomavirus Minor Capsid Protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef] [Green Version]
- Tolstov, Y.L.; Knauer, A.; Chen, J.G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic Primary Merkel Cell Polyomavirus Infection among Adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef] [Green Version]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses Are Chronically Shed from Human Skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of Human Polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Kwun, H.J.; Liu, X.; Gjoerup, O.; Stolz, D.B.; Chang, Y.; Moore, P.S. Cellular and Viral Factors Regulating Merkel Cell Polyomavirus Replication. PLoS ONE 2011, 6, e22468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, Gene Expression and Particle Production by a Consensus Merkel Cell Polyomavirus (MCPyV) Genome. PLoS ONE 2011, 6, e29112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and Sialylated Glycans Sequentially Facilitate Merkel Cell Polyomavirus Infectious Entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef] [Green Version]
- Schowalter, R.M.; Reinhold, W.; Buck, C.B. Entry Tropism of BK and Merkel Cell Polyomaviruses in Cell Culture. PLoS ONE 2012, 7, e42181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Scheller, A.; Prives, C. Simian virus 40 and polyomavirus large tumor antigens have different requirements for high-affinity sequence-specific DNA binding. J. Virol. 1985, 54, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Neu, U.; Hengel, H.; Blaum, B.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel Cell Polyomavirus VP1 Complexes Define a Sialic Acid Binding Site Required for Infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious Entry of Merkel Cell Polyomavirus. J. Virol. 2019, 93, e02004-18. [Google Scholar] [CrossRef] [Green Version]
- Dobson, S.J.; Anene, A.; Boyne, J.; Mankouri, J.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small tumour antigen activates the p38 MAPK pathway to enhance cellular motility. Biochem. J. 2020, 477, 2721–2733. [Google Scholar] [CrossRef]
- Prives, C.; Murakami, Y.; Kern, F.G.; Folk, W.; Basilico, C.; Hurwitz, J. DNA sequence requirements for replication of polyomavirus DNA in vivo and in vitro. Mol. Cell. Biol. 1987, 7, 3694–3704. [Google Scholar]
- Deb, S.; DeLucia, A.L.; Koff, A.; Tsui, S.; Tegtmeyer, P. The adenine-thymine domain of the simian virus 40 core origin directs DNA bending and coordinately regulates DNA replication. Mol. Cell. Biol. 1986, 6, 4578–4584. [Google Scholar]
- Gannon, J.V.; Lane, D. Interactions between SV40 T antigen and DNA polymerase alpha. New Biol. 1990, 2, 84–92. [Google Scholar]
- Onwubiko, N.O.; Borst, A.; Diaz, S.A.; Passkowski, K.; Scheffel, F.; Tessmer, I.; Nasheuer, H.P. SV40 T antigen interactions with ssDNA and replication protein A: A regulatory role of T antigen monomers in lagging strand DNA replication. Nucleic Acids Res. 2020, 48, 3657–3677. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Small T Antigen Controls Viral Replication and Oncoprotein Expression by Targeting the Cellular Ubiquitin Ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.M.; Rio, D.C.; Robbins, A.K.; Tjian, R. SV40 gene expression is modulated by the cooperative binding of T antigen to DNA. Cell 1981, 25, 373–384. [Google Scholar] [CrossRef]
- Rio, D.C.; Tjian, R. SV40 T antigen binding site mutations that affect autoregulation. Cell 1983, 32, 1227–1240. [Google Scholar] [CrossRef]
- Alvarez Orellana, J.; Kwun, H.J.; Artusi, S.; Chang, Y.; Moore, P.S. Sirolimus and Other Mechanistic Target of Rapamycin Inhibitors Directly Activate Latent Pathogenic Human Polyomavirus Replication. J. Infect. Dis. 2021, 224, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Czech-Sioli, M.; Siebels, S.; Radau, S.; Zahedi, R.P.; Schmidt, C.; Dobner, T.; Grundhoff, A.; Fischer, N. The Ubiquitin-Specific Protease Usp7, a Novel Merkel Cell Polyomavirus Large T-Antigen Interaction Partner, Modulates Viral DNA Replication. J. Virol. 2020, 94, e01638-19. [Google Scholar] [CrossRef]
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel Cell Polyomavirus Large T Antigen Disrupts Lysosome Clustering by Translocating Human Vam6p from the Cytoplasm to the Nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, M.A.; He, C.Y.; Chen, Z.Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 53. [Google Scholar] [CrossRef]
- Dye, K.N.; Welcker, M.; Clurman, B.E.; Roman, A.; Galloway, D.A. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and beta-TrCP. PLoS Pathog. 2019, 15, e1007543. [Google Scholar] [CrossRef]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-Positive Merkel Cell Carcinoma Cell Line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Lee, M.S.; Yuan, H.; Jeon, H.; Zhu, Y.; Yoo, S.; Shi, S.; Krueger, B.; Renne, R.; Lu, C.; Jung, J.U.; et al. Human Mesenchymal Stem Cells of Diverse Origins Support Persistent Infection with Kaposi’s Sarcoma-Associated Herpesvirus and Manifest Distinct Angiogenic, Invasive, and Transforming Phenotypes. mBio 2016, 7, e02109-15. [Google Scholar] [CrossRef] [Green Version]
- Porter, S.S.; McBride, A.A. Human Papillomavirus Quasivirus Production and Infection of Primary Human Keratinocytes. Curr. Protoc. Microbiol. 2020, 57, e101. [Google Scholar] [CrossRef]
- Henno, L.; Tombak, E.; Geimanen, J.; Orav, M.; Ustav, E.; Ustav, M. Analysis of Human Papillomavirus Genome Replication Using Two- and Three-Dimensional Agarose Gel Electrophoresis. Curr. Protoc. Microbiol. 2017, 45, 14B.10.1–14B.10.37. [Google Scholar] [CrossRef]
- Guo, X.; Chen, P.; Hou, X.; Xu, W.; Wang, D.; Wang, T.Y.; Zhang, L.; Zheng, G.; Gao, Z.L.; He, C.Y.; et al. The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 2016, 6, 25552. [Google Scholar] [CrossRef] [Green Version]
- Hyde-DeRuyscher, R.P.; Carmichael, G.G. Polyomavirus late pre-mRNA processing: DNA replication-associated changes in leader exon multiplicity suggest a role for leader-to-leader splicing in the early-late switch. J. Virol. 1990, 64, 5823–5832. [Google Scholar] [CrossRef] [Green Version]
- Hyde-DeRuyscher, R.; Carmichael, G.G. Polyomavirus early-late switch is not regulated at the level of transcription initiation and is associated with changes in RNA processing. Proc. Natl. Acad. Sci. USA 1988, 85, 8993–8997. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [Green Version]
- Revie, D.; Tseng, B.Y.; Grafstrom, R.H.; Goulian, M. Covalent association of protein with replicative form DNA of parvovirus H-1. Proc. Natl. Acad. Sci. USA 1979, 76, 5539–5543. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Lin, Y.C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef] [Green Version]
- Adami, G.R.; Marlor, C.W.; Barrett, N.L.; Carmichael, G.G. Leader-to-leader splicing is required for efficient production and accumulation of polyomavirus late mRNAs. J. Virol. 1989, 63, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Bayer, N.J.; Januliene, D.; Zocher, G.; Stehle, T.; Moeller, A.; Blaum, B.S. Structure of Merkel Cell Polyomavirus Capsid and Interaction with Its Glycosaminoglycan Attachment Receptor. J. Virol. 2020, 94, e01664-19. [Google Scholar] [CrossRef]
- Krump, N.A.; Wang, R.; Liu, W.; Yang, J.F.; Ma, T.; You, J. Merkel Cell Polyomavirus Infection Induces an Antiviral Innate Immune Response in Human Dermal Fibroblasts. J. Virol. 2021, 95, 0221120. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final Constructs | Mutation Sites | Insert Source | Restriction Enzymes |
|---|---|---|---|
| pMC-MCV-Rep– | C44A | pJ-MCV-HF-Rep– | AvrII, SacI |
| pMC-MCV-hVam6p– | T1251G, G1252C | pJ-MCV-HF-hVam6p– | BamHI, EcoRI |
| pMC-MCV-β-TrCP– | T1065G | pJ-MCV-HF-β-TrCP– | AvrII, BamHI |
| pMC-MCV-Skp2– | T1284G | pJ-MCV-HF-Skp2– | BamHI, EcoRI |
| pMC-MCV-Fbw7– | T1341G | pJ-MCV-HF-Fbw7– | BamHI, EcoRI |
| VP1 (2440-2466) F | TGACACATTGCAGATGTGGGAGGCAAT |
| VP1-mScarlet R | GCCTCGCCCTTGCTCACCATTAATTCTTGTGTTTGGCTTT |
| VP1-mScarlet F | AAAGCCAAACACAAGAATTAATGGTGAGCAAGGGCGAGGC |
| mScarlet-attBVec R | TCCCCGGGCGCGACAAATAATTCTCACTTGTACAGCTCGT |
| VP1-P2A R | GTCTCCAGCCTGCTTCAGCAGGCTGAAGTTAGTAGCTCCGCTTCCTAATTCTTGTGTTTGGCTTTCTTTTTGAGAGGCC |
| P2A-mScarlet F | AGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTATGGTGAGCAAGGGCGAGGCA |
| VP1-P2A_FW | GCCAAGCTTGCATGCCGTTCTTTAATTAATGTTCATTATT |
| VP1-P2A_RV | CCAGCCTGCTTCAGCAGGCTGAAGTTAGTAGC |
| P2A-mScarlet_FW | TGCTGAAGCAGGCTGGAGACGTGGAGGAGAAC |
| P2A-mScarlet_RV | AATTCGAGCTCGGTACTCCCCGGGCGCGACAAATAATTCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abere, B.; Zhou, H.; Shuda, M.; Stolz, D.B.; Rapchak, K.; Moore, P.S.; Chang, Y. Replication Kinetics for a Reporter Merkel Cell Polyomavirus. Viruses 2022, 14, 473. https://doi.org/10.3390/v14030473
Abere B, Zhou H, Shuda M, Stolz DB, Rapchak K, Moore PS, Chang Y. Replication Kinetics for a Reporter Merkel Cell Polyomavirus. Viruses. 2022; 14(3):473. https://doi.org/10.3390/v14030473
Chicago/Turabian StyleAbere, Bizunesh, Hongzhao Zhou, Masahiro Shuda, Donna B. Stolz, Kyle Rapchak, Patrick S. Moore, and Yuan Chang. 2022. "Replication Kinetics for a Reporter Merkel Cell Polyomavirus" Viruses 14, no. 3: 473. https://doi.org/10.3390/v14030473
APA StyleAbere, B., Zhou, H., Shuda, M., Stolz, D. B., Rapchak, K., Moore, P. S., & Chang, Y. (2022). Replication Kinetics for a Reporter Merkel Cell Polyomavirus. Viruses, 14(3), 473. https://doi.org/10.3390/v14030473