
Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Maximum Likelihood Phylogenetic Analysis
2.2. Mutations and Glycosylation Pattern
2.3. SARS-CoV-2 Gene Flows, Evolutionary Rate Estimate, and Time-Scaled Phylogeny among Italian Regions
2.4. Ethical Approval
3. Results
3.1. Phylogenetic Assignment through Pangolin COVID-19 Lineage Assigner and Maximum Likelihood Phylogenetic Analysis
3.2. Mutations and Glycosylation Pattern
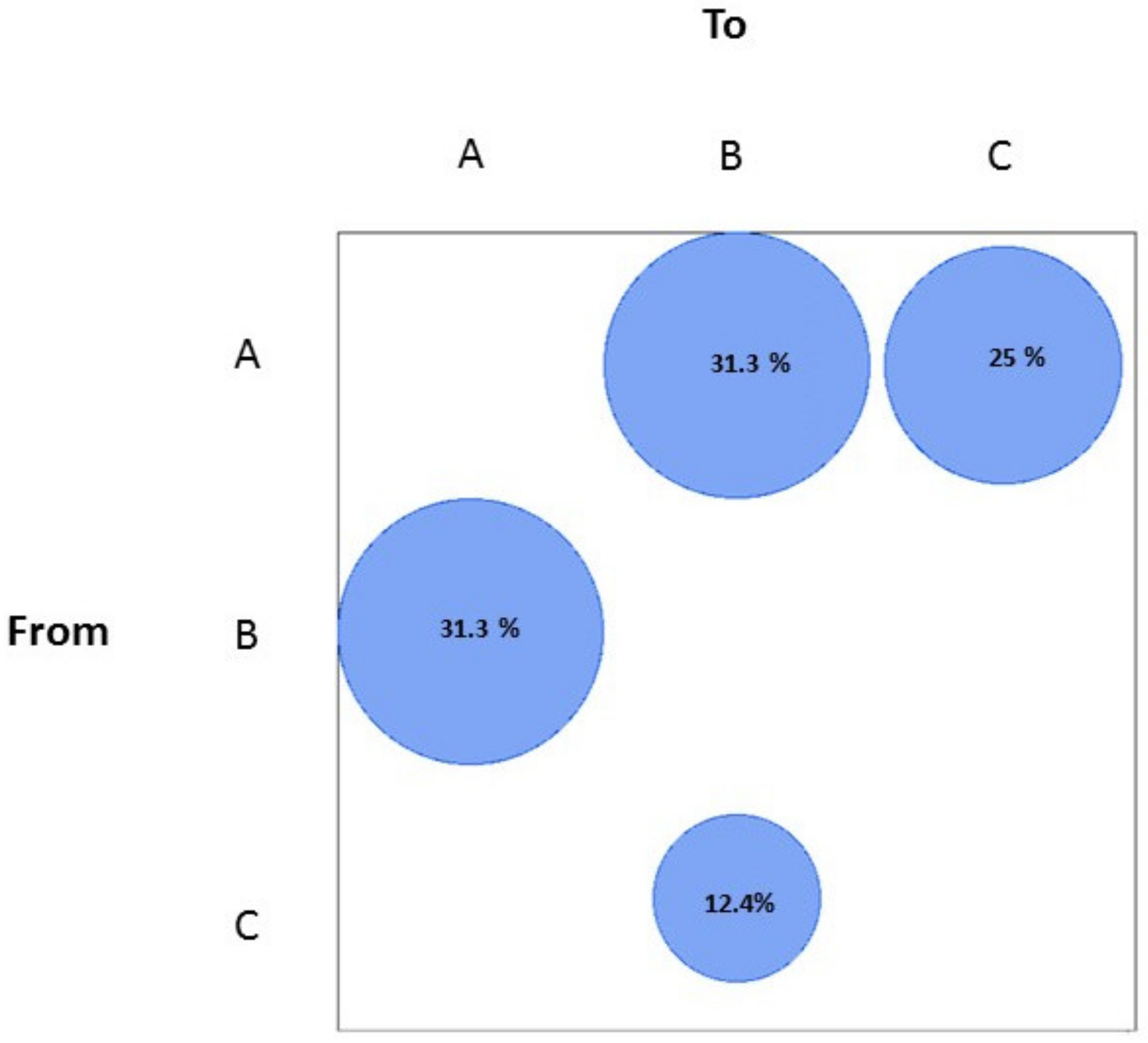
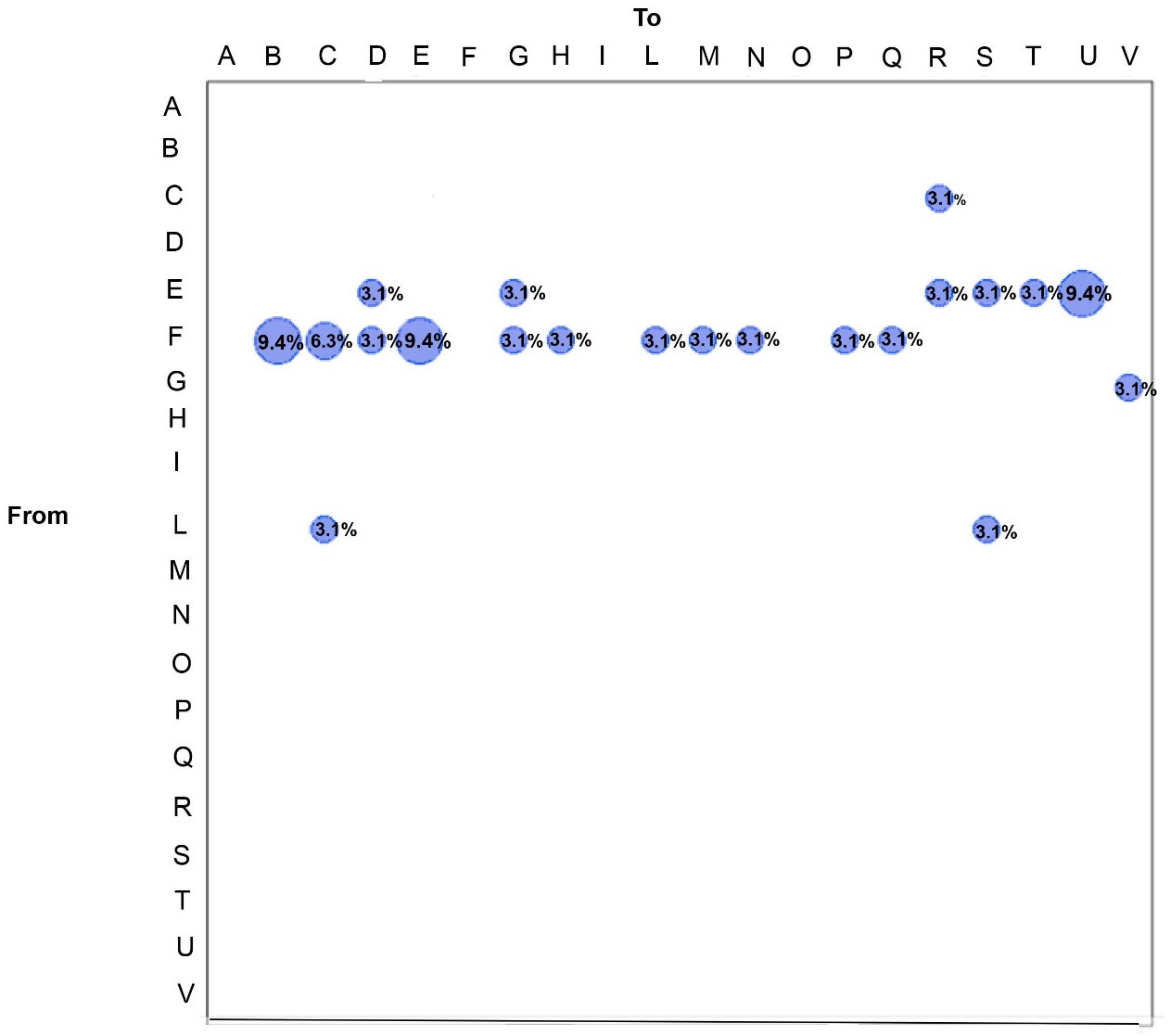
3.3. SARS-CoV-2 Gene Flows in Italy
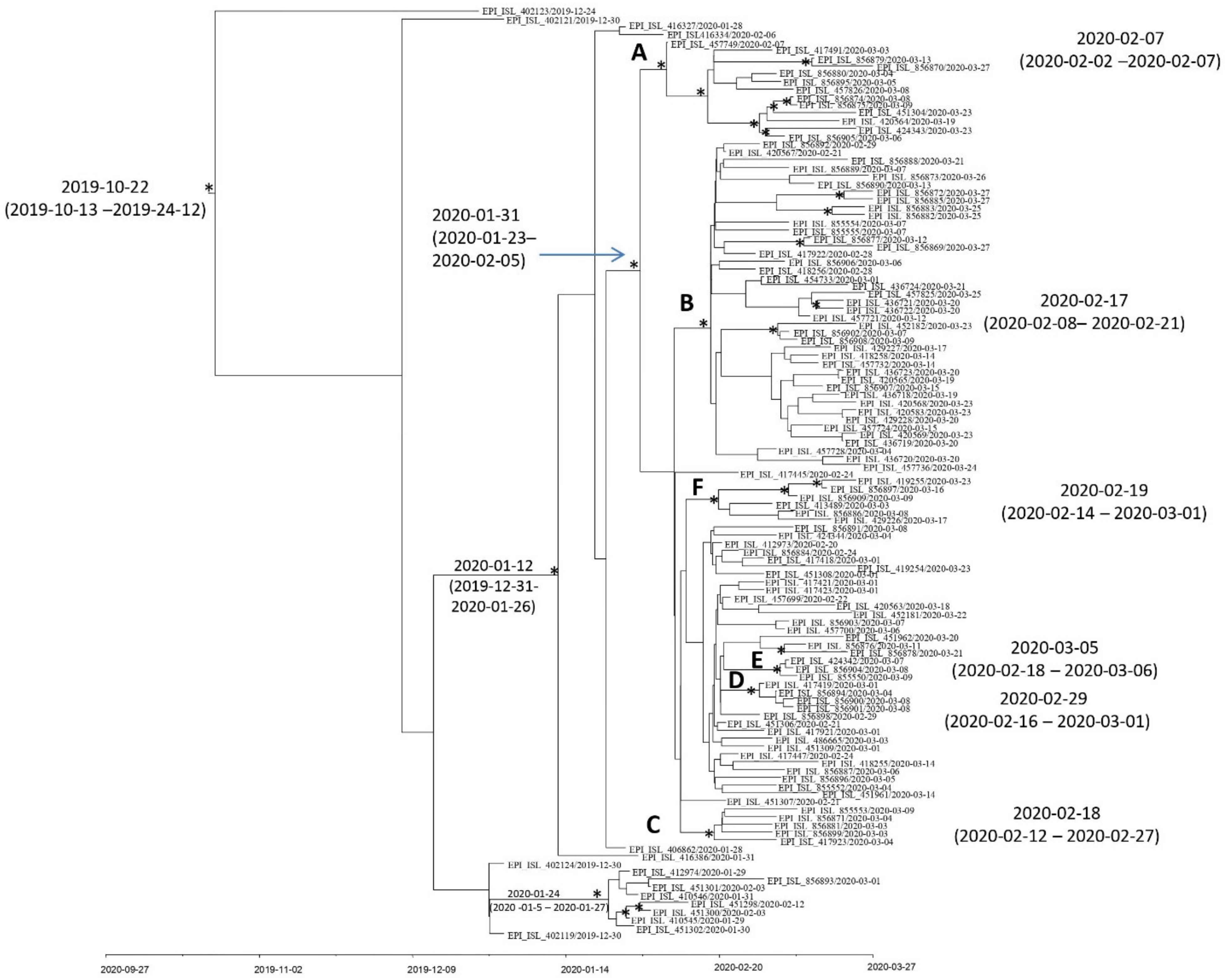
3.4. Evolutionary Rate Estimate and Time-Scaled Phylogeny
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Statement on the Second Meeting of the International Health Regulations (2005) Emergency Committee Regarding the Outbreak of Novel Coronavirus (2019-nCoV). 30 January 2020. Available online: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 29 September 2021).
- World Health Organization (WHO). WHO Director—General’s Opening Remarks at the Media Briefing on COVID-19. 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 29 September 2021).
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F.; et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 954–956. [Google Scholar] [CrossRef]
- Stefanelli, P.; Faggioni, G.; Lo Presti, A.; Fiore, F.; Marchi, M.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. On behalf of ISS COVID-19 study group. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Eurosurveillance 2020, 25, 2000305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EpiCentro—L’Epidemiologia per la Sanità Pubblica—Istituto Superiore di Sanità—Sorveglianza Integrata COVID-19. Available online: https://www.epicentro.iss.it/coronavirus/sars-cov-2-sorveglianza-dati (accessed on 29 September 2021).
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, L.; Bianco, A.; Del Sambro, L.; Simone, D.; Lippolis, A.; Notarnicola, M.; Pesole, G.; Pace, L.; Galante, D.; Parisi, A. Genomic Surveillance of Circulating SARS-CoV-2 in South East Italy: A One-Year Retrospective Genetic Study. Viruses 2021, 13, 731. [Google Scholar] [CrossRef]
- Di Giallonardo, F.; Duchene, S.; Puglia, I.; Curini, V.; Profeta, F.; Cammà, C.; Marcacci, M.; Calistri, P.; Holmes, E.C.; Lorusso, A. Genomic Epidemiology of the First Wave of SARS-CoV-2 in Italy. Viruses 2020, 12, 1438. [Google Scholar] [CrossRef]
- GISAID. Available online: https://www.gisaid.org/ (accessed on 15 June 2020).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Pangolin Lineage Assigner. Version 3.1.14, Lineage Version. Available online: https://cov-lineages.org/lineage_list.html (accessed on 13 October 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; Mc Crone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- IQ-TREE. Available online: http://www.iqtree.org (accessed on 23 June 2020).
- Trifinopoulos, J.; Nguyen, L.-T.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 25 June 2020).
- Zhang, M.; Gaschen, B.; Blay, W.; Foley, B.; Haigwood, N. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 2004, 14, 1229–1246. [Google Scholar] [CrossRef] [Green Version]
- N-Glycosite. Available online: https://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html (accessed on 14 September 2020).
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef]
- Maddison, D.R.; Maddison, W.P. MacClade 4: Analysis of Phylogeny and Character Evolution; Version 4.0.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Slatkin, M.; Maddison, W.P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 1989, 123, 603–613. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes Factor. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Vansteelandt, S. Make the most of your samples: Bayes factor estimators for high-dimensional models of sequence evolution. BMC Bioinform. 2013, 14, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A. Phylodynamic Analysis. 2020. Available online: http://virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356 (accessed on 6 March 2020).
- Zehender, G.; Lai, A.; Bergna, A.; Meroni, L.; Riva, A.; Balotta, C.; Tarkowski, M.; Gabrieli, A.; Bernacchia, D.; Rusconi, S.; et al. Genomic characterization and phylogenetic analysis of SARS-COV-2 in Italy. J. Med. Virol. 2020, 92, 1637–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; Bergna, A.; Acciarri, C.; Galli, M.; Zehender, G. Early phylogenetic estimate of the effective reproduction number of SARS-CoV-2. J. Med. Virol. 2020, 92, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Lo Presti, A.; Rezza, G.; Stefanelli, P. Selective pressure on SARS-CoV-2 protein coding genes and glycosylation site prediction. Heliyon 2020, 6, e05001. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Chand, G.B.; Banerjee, A.; Azad, G.K. Identification of novel mutations in RNA-dependent RNA polymerases of SARS-CoV-2 and their implications on its protein structure. PeerJ 2020, 8, e9492. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. SARS-CoV-2 Spike protein variant D614G increases infectivity and retains sensitivity to antibodies that target the receptor binding domain. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19. Virus Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Rahman, M.S.; Islam, M.R.; Alam, A.S.M.R.U.; Islam, I.; Hoque, M.N.; Akter, S.; Rahaman, M.; Sultana, M.; Hossain, M.A. Evolutionary dynamics of SARS-CoV-2 nucleocapsid protein and its consequences. J. Med. Virol. 2020, 93, 2177–2195. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.W. Decoding asymptomatic COVID-19 infection and transmission. J. Phys. Chem. Lett. 2020, 11, 10007–10015. [Google Scholar] [CrossRef]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y. Effects of SARS-CoV-2 mutations on protein structures and intraviral protein–protein interactions. J. Med. Virol. 2020, 93, 2132–2140. [Google Scholar] [CrossRef]
- Micheli, V.; Rimoldi, S.G.; Romeri, F.; Comandatore, F.; Mancon, A.; Gigantiello, A.; Perini, M.; Mileto, D.; Pagani, C.; Lombardi, A.; et al. Geographical reconstruction of the SARS-CoV-2 outbreak in Lombardy (Italy) during the early phase. J. Med. Virol. 2020, 93, 1752–1757. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Pontremoli, C.; Mozzi, A.; Pozzoli, U.; Clerici, M.; Sironi, M. Antigenic variation of SARS-CoV-2 in response to immune pressure. Mol. Ecol. 2020, 00, 1–12. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Rutkowska, M.; Poston, D.; Da Silva, J.; Zhang, F.; Bednarski, E.; Cho, A.; Schaefer-Babajew, D.J.; Gaebler, C.; et al. High genetic barrier to escape from human polyclonal SARS-CoV-2 neutralizing antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, E.; Renzetti, S.; Volta, M.; Donato, F.; Calza, S.; Placidi, D.; Lucchini, R.G.; Rota, M. COVID-19 incidence and mortality in Lombardy, Italy: An ecological study on the role of air pollution, meteorological factors, demographic and socioeconomic variables. Environ. Res. 2021, 195, 110777. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef] [Green Version]
- Spiteri, G.; Fielding, J.; Diercke, M.; Campese, C.; Enouf, V.; Gaymard, A.; Bella, A.; Sognamiglio, P.; Moros, M.J.S.; Riutort, A.N.; et al. First cases of coronavirus disease 2019 (COVID-19) in the WHO European Region, 24 January to 21 February 2020. Eurosurveillance 2020, 25, 2000178. [Google Scholar] [CrossRef] [Green Version]
- Faggioni, G.; Stefanelli, P.; Giordani, F.; Fillo, S.; Anselmo, A.; Fain, V.V.; Fortunato, A.; Petralito, G.; Molinari, F.; Lo Presti, A.; et al. Identification and characterization of SARS-CoV-2 clusters in the EU/EEA in the first pandemic wave: Additional elements to trace the route of the virus. Infect. Genet. Evol. 2021, 96, 105108. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | Cluster | Lineage | Total | Region and Number |
|---|---|---|---|---|
| main | a | B.1 | 6 | Umbria (1), Lazio (4), Abruzzo (1) |
| main | b | B.1 | 2 | Campania (1), Lombardy (1) |
| sub-clade | c | B.1 | 6 | Veneto (1), Molise (1), Abruzzo (2), Lazio (1), Lombardy (1) |
| sub-clade | d | B.1 | 3 | Lombardy (1), Lazio (1), Tuscany (1) |
| sub-clade | e | B.1 | 4 | Sicily (2), Friuli-Venezia Giulia (2) |
| sub-clade | f | B.1 | 2 | Marche (1), Lombardy (1) |
| sub-clade | g | B.1 | 2 | Campania (1), Apulia (1) |
| sub-clade | h | B.1 | 5 | Piedmont (1), Molise (1), Lombardy (1), Campania (1), Lazio (1) |
| sub-clade | i | B.1 | 2 | Lazio (2) |
| sub-clade | l | B.1.1 (39), B.1.1.323 (1) | 42 | Abruzzo (16), Tuscany (2), Veneto (2), AP of Bolzano (1) |
| Lazio (3), Campania (2), Calabria (1), Lombardy (3), Sardinia (3), Valle d’Aosta (3), Umbria (1), Sicily (1), Basilicata (2) | ||||
| B.1.1.61 (1) | Friuli-Venezia Giulia (1), Molise (1). | |||
| B.1.1.70 (1) |
| Mutation | n/Total | Percentage | n/58 (ISS and Army Medical Center) | Percentage | Target | Gene |
|---|---|---|---|---|---|---|
| P323L | 103/111 | 92.80% | 56/58 | 96.50% | nsp12-RNA-dependent RNA polymerase | ORF1ab |
| D614G | 103/111 | 92.80% | 56/58 | 96.50% | spike | S |
| R203K | 41/111 | 37% | 23/58 | 39.70% | nucleocapsid | N |
| G204R | 41/111 | 37% | 23/58 | 39.70% | nucleocapsid | N |
| D3G | 13/111 | 11.70% | 9/58 | 15.50% | membrane | M |
| L37F | 9/111 | 8.10% | 4/58 | 6.90% | nsp6 | ORF1ab |
| G251V | 8/111 | 7.20% | 2/58 | 3.44% | ORF3a | ORF3a |
| V246I | 4/111 | 3.60% | 3/58 | 5.20% | nucleocapsid | N |
| T175M | 3/111 | 2.70% | 2/58 | 3.44% | membrane | M |
| L5F | 3/111 | 2.70% | 2/58 | 3.44% | spike | S |
| A302V | 2/111 | 1.80% | 2/58 | 3.44% | nsp2 | ORF1ab |
| D218E | 2/111 | 1.80% | 2/58 | 3.44% | nsp3 | ORF1ab |
| S74A | 2/111 | 1.80% | 1/58 | 1.72% | nsp13/helicase | ORF1ab |
| A81V | 2/111 | 1.80% | 2/58 | 3.44% | nsp 15/endoRNAse | ORF1ab |
| Q218R | 2/111 | 1.80% | 2/58 | 3.44% | nsp16/2’-O-ribose methyltransferase | ORF1ab |
| L41F | 2/111 | 1.80% | 2/58 | 3.44% | ORF3a | ORF3a |
| H182R | 2/111 | 1.80% | 2/58 | 3.44% | ORF3a | ORF3a |
| Q12 * | 2/111 | 1.80% | 2/58 | 3.44% | ORF10 | ORF10 |
| P191S | 1/111 | 0.90% | 1/58 | 1.72% | nsp2 | ORF1ab |
| T237I | 1/111 | 0.90% | 1/58 | 1.72% | nsp3 | ORF1ab |
| A496V | 1/111 | 0.90% | 1/58 | 1.72% | nsp3 | ORF1ab |
| A534V | 1/111 | 0.90% | 1/58 | 1.72% | nsp3 | ORF1ab |
| P1159S | 1/111 | 0.90% | / | / | nsp3 | ORF1ab |
| S1285F | 1/111 | 0.90% | / | / | nsp3 | ORF1ab |
| I1413L | 1/111 | 0.90% | / | / | nsp3 | ORF1ab |
| M1547I | 1/111 | 0.90% | / | / | nsp3 | ORF1ab |
| N1785D | 1/111 | 0.90% | 1/58 | 1.72% | nsp3 | ORF1ab |
| A1872V | 1/111 | 0.90% | / | / | nsp3 | ORF1ab |
| V357I | 1/111 | 0.90% | 1/58 | 1.72% | nsp4 | ORF1ab |
| A457V | 1/111 | 0.90% | 1/58 | 1.72% | nsp4 | ORF1ab |
| K285N | 1/111 | 0.90% | / | / | nsp6 | ORF1ab |
| A287S | 1/111 | 0.90% | / | / | nsp6 | ORF1ab |
| S10L | 1/111 | 0.90% | / | / | nsp7 | ORF1ab |
| F49I | 1/111 | 0.90% | / | / | nsp7 | ORF1ab |
| Y149 * | 1/111 | 0.90% | / | / | nsp8 | ORF1ab |
| W154R | 1/111 | 0.90% | / | / | nsp8 | ORF1ab |
| P80S | 1/111 | 0.90% | 1/58 | 1.72% | nsp9 | ORF1ab |
| R285H | 1/111 | 0.90% | 1/58 | 1.72% | nsp12-RNA-dependent RNA polymerase | ORF1ab |
| S363R | 1/111 | 0.90% | 1/58 | 1.72% | nsp12-RNA-dependent RNA polymerase | ORF1ab |
| D893Y | 1/111 | 0.90% | / | / | nsp12-RNA-dependent RNA polymerase | ORF1ab |
| P529L | 1/111 | 0.90% | 1/58 | 1.72% | nsp13/helicase | ORF1ab |
| T31I | 1/111 | 0.90% | 1/58 | 1.72% | nsp14/3’-to-5’ exonuclease | ORF1ab |
| V287F | 1/111 | 0.90% | 1/58 | 1.72% | nsp14/3’-to-5’ exonuclease | ORF1ab |
| P23S | 1/111 | 0.90% | / | / | nsp 15/endoRNAse | ORF1ab |
| S154F | 1/111 | 0.90% | / | / | nsp 15/endoRNAse | ORF1ab |
| K160R | 1/111 | 0.90% | 1/58 | 1.72% | nsp16/2’-O-ribose methyltransferase | ORF1ab |
| A222S | 1/111 | 0.90% | 1/58 | 1.72% | spike | S |
| P681S | 1/111 | 0.90% | 1/58 | 1.72% | spike | S |
| D839Y | 1/111 | 0.90% | / | / | spike | S |
| M1T | 1/111 | 0.90% | 1/58 | 1.72% | ORF3a | ORF3a |
| D2Y | 1/111 | 0.90% | 1/58 | 1.72% | ORF3a | ORF3a |
| Q57H | 1/111 | 0.90% | 1/58 | 1.72% | ORF3a | ORF3a |
| A110S | 1/111 | 0.90% | / | / | ORF3a | ORF3a |
| G6V | 1/111 | 0.90% | 1/58 | 1.72% | membrane | M |
| W29R | 1/111 | 0.90% | 1/58 | 1.72% | ORF7b | ORF7b |
| D22Y | 1/111 | 0.90% | 1/58 | 1.72% | nucleocapsid | N |
| T24N | 1/111 | 0.90% | 1/58 | 1.72% | nucleocapsid | N |
| D103Y | 1/111 | 0.90% | / | / | nucleocapsid | N |
| R185H | 1/111 | 0.90% | 1/58 | 1.72% | nucleocapsid | N |
| L331F | 1/111 | 0.90% | 1/58 | 1.72% | nucleocapsid | N |
| A381V | 1/111 | 0.90% | 1/58 | 1.72% | nucleocapsid | N |
| G50N | 41/111 | 37% | 23/58 | 39.7% | ORF14 (uncharacterized protein) | ORF14 |
| G50E | 1/111 | 0.90% | 1/58 | 1.72% | ORF14 (uncharacterized protein) | ORF14 |
| V32I | 1/111 | 0.90% | 1/58 | 1.72% | ORF14 (uncharacterized protein) | ORF14 |
| R32L | 1/111 | 0.90% | 1/58 | 1.72% | ORF9b | ORF9b |
| L21M | 1/111 | 0.90% | 1/58 | 1.72% | ORF9b | ORF9b |
| Q18H | 1/111 | 0.90% | 1/58 | 1.72% | ORF9b | ORF9b |
| G71S | 1/111 | 0.90% | 1/58 | 1.72% | nsp5 | ORF1ab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Presti, A.; Di Martino, A.; Faggioni, G.; Giordani, F.; Fillo, S.; Anselmo, A.; Fain, V.V.; Fortunato, A.; Petralito, G.; Molinari, F.; et al. Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses 2022, 14, 472. https://doi.org/10.3390/v14030472
Lo Presti A, Di Martino A, Faggioni G, Giordani F, Fillo S, Anselmo A, Fain VV, Fortunato A, Petralito G, Molinari F, et al. Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses. 2022; 14(3):472. https://doi.org/10.3390/v14030472
Chicago/Turabian StyleLo Presti, Alessandra, Angela Di Martino, Giovanni Faggioni, Francesco Giordani, Silvia Fillo, Anna Anselmo, Vanessa Vera Fain, Antonella Fortunato, Giancarlo Petralito, Filippo Molinari, and et al. 2022. "Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020" Viruses 14, no. 3: 472. https://doi.org/10.3390/v14030472
APA StyleLo Presti, A., Di Martino, A., Faggioni, G., Giordani, F., Fillo, S., Anselmo, A., Fain, V. V., Fortunato, A., Petralito, G., Molinari, F., Palomba, S., De Santis, R., Fiore, S., Fabiani, C., Di Mario, G., Facchini, M., Calzoletti, L., Lista, F., Rezza, G., & Stefanelli, P. (2022). Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses, 14(3), 472. https://doi.org/10.3390/v14030472