
Virus-like Particles: Measures and Biological Functions




Abstract
1. Introduction
2. Counting Particles
2.1. Virus and Virus-like Particles
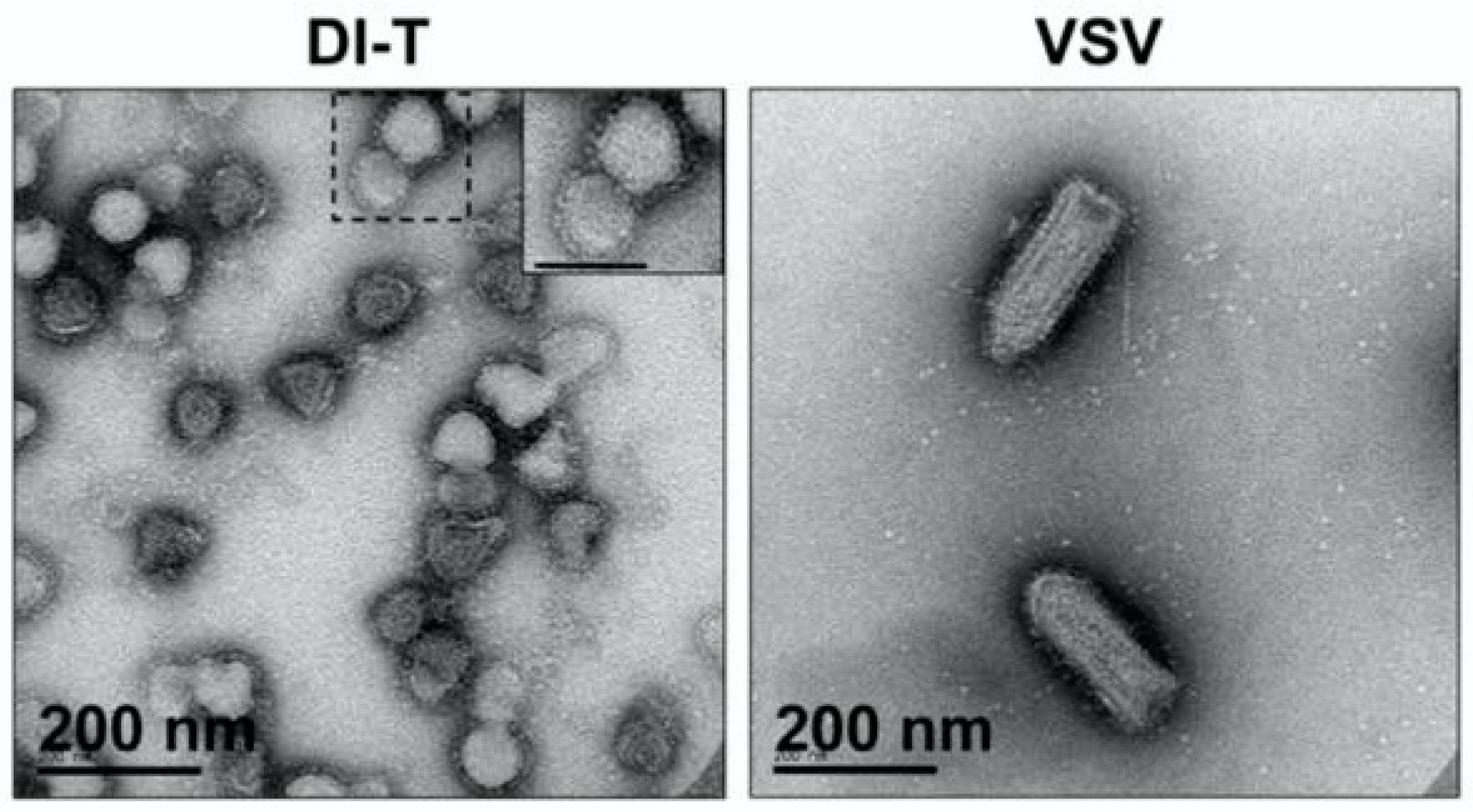
2.1.1. Transmission Electron Microscopy
2.1.2. Epifluorescence Microscopy
2.1.3. Resistive Pulse Sensing
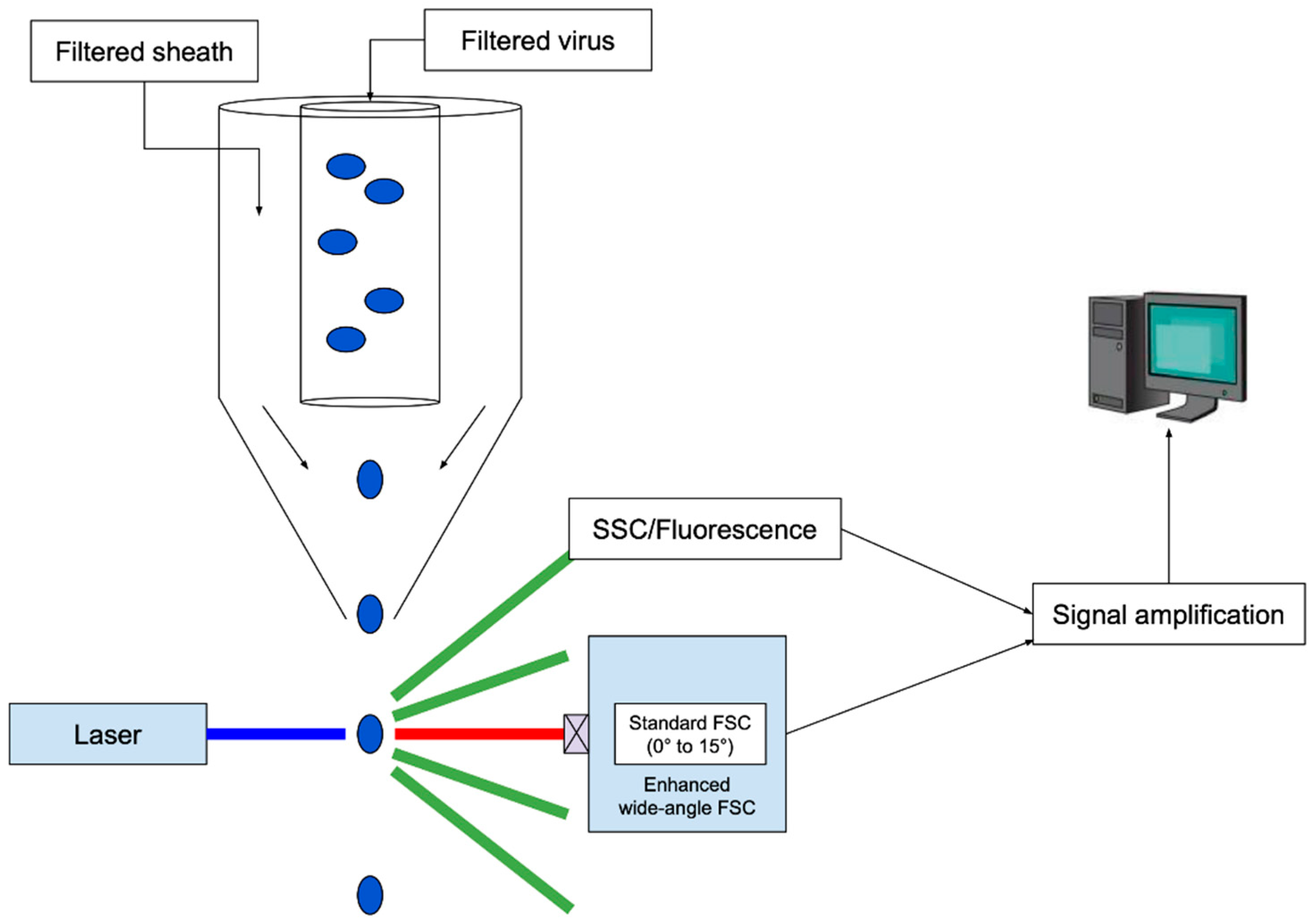
2.1.4. Flow Cytometry and Virometry
2.2. Infectious Virus Particles
2.2.1. Plaque Assay
2.2.2. End-Point Dilution
2.3. Cell-Killing Particles (Clonogenic Assay)
3. Virus-like Particles: Emergence, Function, and Prevalence
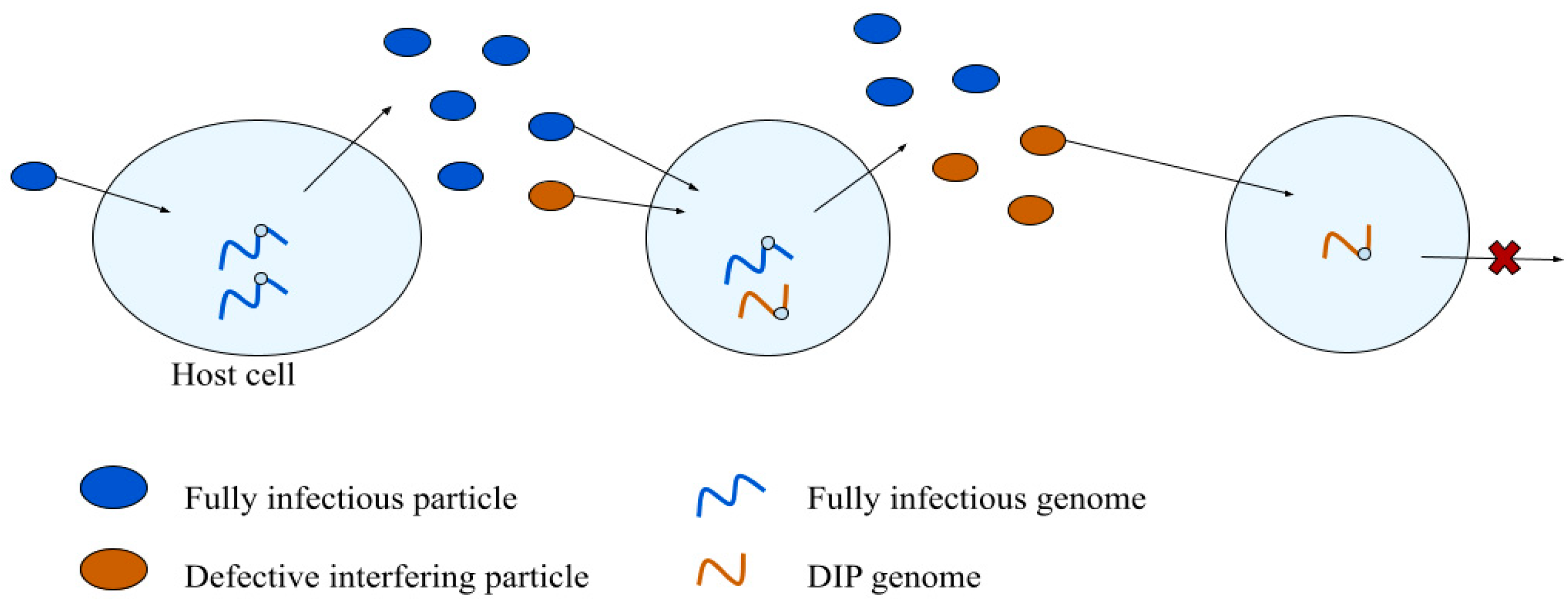
3.1. Defective Interfering Particles
3.1.1. DIP Emergence
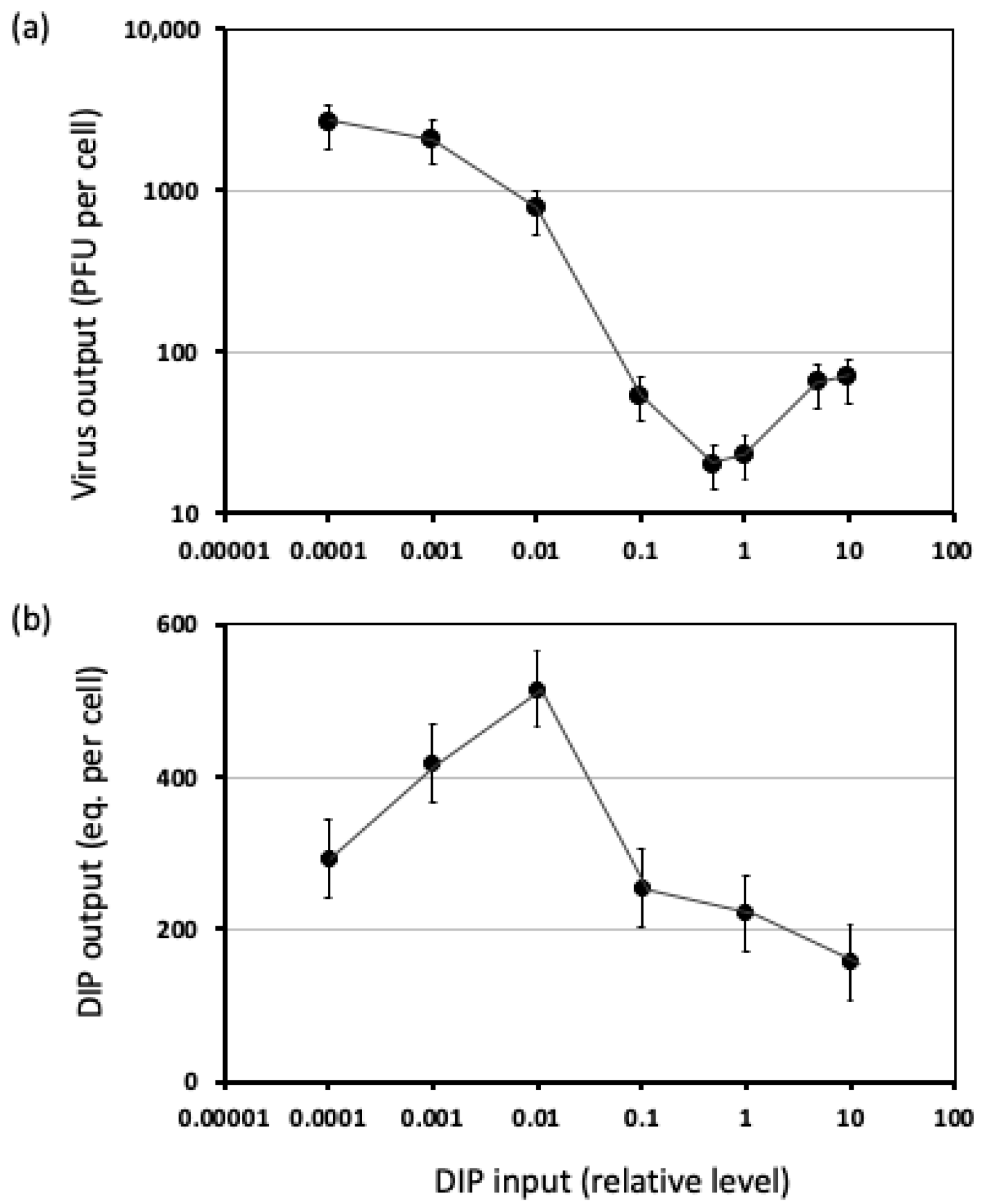
3.1.2. Measures of DIP Interference
3.1.3. DIPs In Vitro
3.1.4. DIPs In Vivo
3.1.5. DIPs and the Immune Response
3.1.6. DIPs as Antiviral Therapies
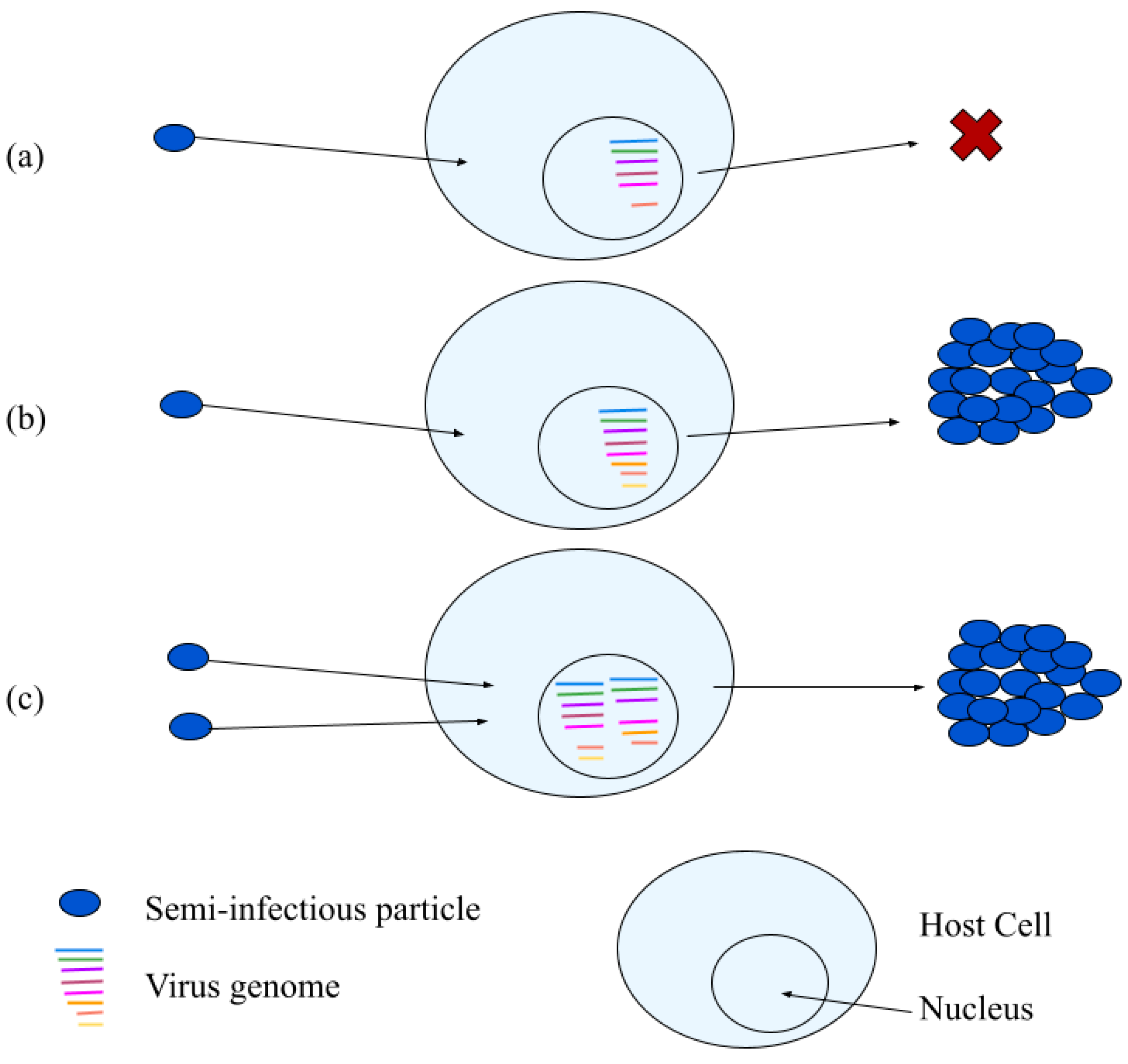
3.2. Semi-Infectious Particles
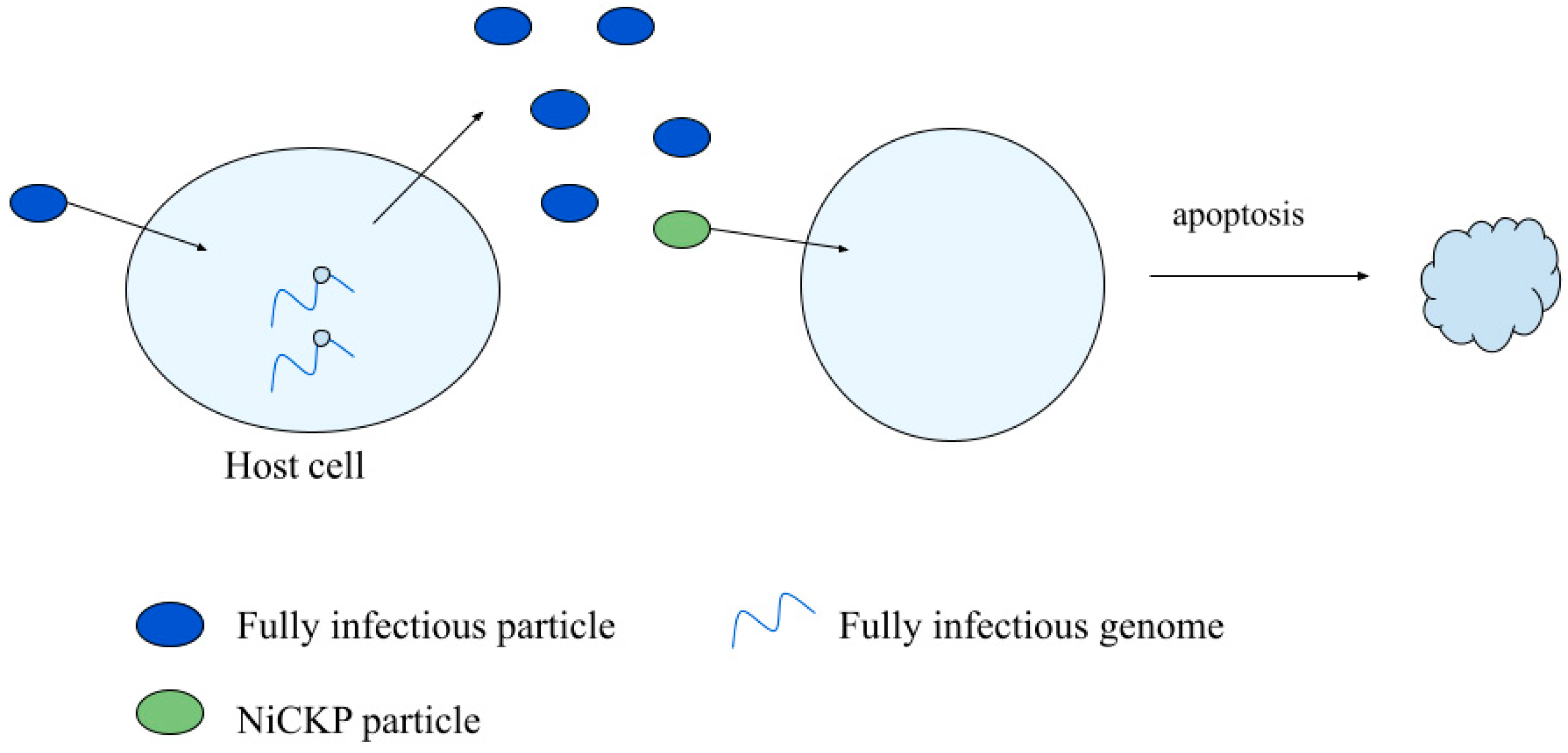
3.3. Non-Infectious Cell Killing Particles
3.3.1. NiCKP Characterization
3.3.2. Particle Fitness and Virulence
3.3.3. Application of NiCKPs
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Stephenson, B. Epitome of Copernican Astronomy. In Kepler’s Physical Astronomy; Springer: New York, NY, USA, 1987; pp. 138–201. [Google Scholar]
- Darwin, C. On the Origin of Species, 1859; Routledge: Oxford, UK, 2004. [Google Scholar]
- Burnet, F. Immunological studies with the virus of infectious laryngotracheitis of fowls using the developing egg technique. J. Exp. Med. 1936, 63, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.L.; Delbrück, M. The growth of bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Von Borries, B.; Ruska, E.; Ruska, H. Bakterien und Virus in übermikroskopischer Aufnahme. Klin. Wochenschr. 1938, 17, 921–925. [Google Scholar] [CrossRef]
- Overman, J.R.; Tamm, I. Equivalence between vaccinia particles counted by electron microscopy and infectious units of the virus. Proc. Soc. Exp. Biol. Med. 1956, 92, 806–810. [Google Scholar] [CrossRef]
- Watson, D.; Russell, W.; Wildy, P. Electron microscopic particle counts on herpes virus using the phosphotungstate negative staining technique. Virology 1963, 19, 250–260. [Google Scholar] [CrossRef]
- Flint, S.; Enquist, L.; Racaniello, V.; Skalka, A. Principles of Virology: Molecular Biology, Pathogenesis, and Control of Animal Viruses, 2nd ed.; ASM Press: Washington, DC, USA, 2004. [Google Scholar]
- Carpenter, J.E.; Henderson, E.P.; Grose, C. Enumeration of an extremely high particle-to-PFU ratio for varicella-zoster virus. J. Virol. 2009, 83, 6917–6921. [Google Scholar] [CrossRef]
- Ghezzi, S.; Pagani, I.; Poli, G.; Pal, S.; Licciulli, A.; Perboni, S.; Vicenzi, E. Rapid Inactivation of SARS-CoV-2 by Coupling Tungsten Trioxide (WO3) Photocatalyst with Copper Nanoclusters. J. Nanotechnol. Nanomater. 2020, 1, 109–115. [Google Scholar]
- Klimstra, W.B.; Tilston-Lunel, N.L.; Nambulli, S.; Boslett, J.; McMillen, C.M.; Gilliland, T.; Dunn, M.D.; Sun, C.; Wheeler, S.E.; Wells, A. SARS-CoV-2 growth, furin-cleavage-site adaptation and neutralization using serum from acutely infected hospitalized COVID-19 patients. J. Gen. Virol. 2020, 101, 1156–1169. [Google Scholar] [CrossRef]
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formulation for clinical translation. Expert Rev. Vaccines 2018, 17, 833–849. [Google Scholar] [CrossRef]
- Hill, B.D.; Zak, A.; Khera, E.; Wen, F. Engineering virus-like particles for antigen and drug delivery. Curr. Protein Pept. Sci. 2018, 19, 112–127. [Google Scholar] [CrossRef]
- Timm, C.; Akpinar, F.; Yin, J. Quantitative characterization of defective virus emergence by deep sequencing. J. Virol. 2014, 88, 2623. [Google Scholar] [CrossRef] [PubMed]
- Blancett, C.D.; Fetterer, D.P.; Koistinen, K.A.; Morazzani, E.M.; Monninger, M.K.; Piper, A.E.; Kuehl, K.A.; Kearney, B.J.; Norris, S.L.; Rossi, C.A. Accurate virus quantitation using a Scanning Transmission Electron Microscopy (STEM) detector in a scanning electron microscope. J. Virol. Methods 2017, 248, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P.; Raynal, P.I.; Eymieux, S.; Blanchard, E. Virus detection by transmission electron microscopy: Still useful for diagnosis and a plus for biosafety. Rev. Med. Virol. 2019, 29, e2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Lu, J.-R.; Binder, B.J.; Liu, Y.-C.; Hodson, R.E. Application of digital image analysis and flow cytometry to enumerate marine viruses stained with SYBR Gold. Appl. Environ. Microbiol. 2001, 67, 539–545. [Google Scholar] [CrossRef]
- Ortmann, A.C.; Suttle, C.A. Determination of virus abundance by epifluorescence microscopy. In Bacteriophages. Methods in Molecular Biology; Clokie, M.R., Kropinski, A.M., Eds.; Springer: Cham, Switzerland, 2009; Volume 501, pp. 87–95. [Google Scholar]
- Parveen, N.; Borrenberghs, D.; Rocha, S.; Hendrix, J. Single viruses on the fluorescence microscope: Imaging molecular mobility, interactions and structure sheds new light on viral replication. Viruses 2018, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yamamoto, T. Quantification of virus particles using nanopore-based resistive-pulse sensing techniques. Front. Microbiol. 2016, 7, 1500. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, F.; Yin, J. Characterization of vesicular stomatitis virus populations by tunable resistive pulse sensing. J. Virol. Methods 2015, 218, 71–76. [Google Scholar] [CrossRef]
- Rossi, C.A.; Kearney, B.J.; Olschner, S.P.; Williams, P.L.; Robinson, C.G.; Heinrich, M.L.; Zovanyi, A.M.; Ingram, M.F.; Norwood, D.A.; Schoepp, R.J. Evaluation of ViroCyt® Virus Counter for rapid filovirus quantitation. Viruses 2015, 7, 857–872. [Google Scholar] [CrossRef]
- Zamora, J.L.R.; Aguilar, H.C. Flow virometry as a tool to study viruses. Methods 2018, 134, 87–97. [Google Scholar] [CrossRef]
- Nagler, F.; Rake, G. The use of the electron microscope in diagnosis of variola, vaccinia, and varicella. J. Bacteriol. 1948, 55, 45–51. [Google Scholar] [CrossRef]
- Paddock, C.D.; Nicholson, W.L.; Bhatnagar, J.; Goldsmith, C.S.; Greer, P.W.; Hayes, E.B.; Risko, J.A.; Henderson, C.; Blackmore, C.G.; Lanciotti, R.S.; et al. Fatal hemorrhagic fever caused by West Nile virus in the United States. Clin. Infect. Dis. 2006, 42, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Potdar, V.; Cherian, S.; Abraham, P.; Basu, A.; Team, I.C. Transmission electron microscopy imaging of SARS-CoV-2. Indian J. Med. Res. 2020, 151, 241. [Google Scholar] [PubMed]
- Smadel, J.E.; Rivers, T.M.; Pickels, E.G. Estimation of the purity of preparations of elementary bodies of vaccinia. J. Exp. Med. 1939, 70, 379. [Google Scholar] [CrossRef] [PubMed]
- Malenovska, H. Virus quantitation by transmission electron microscopy, TCID50, and the role of timing virus harvesting: A case study of three animal viruses. J. Virol. Methods 2013, 191, 136–140. [Google Scholar] [CrossRef]
- Reid, G.; Milne, E.; Coggins, L.; Wilson, N.; Smith, K.; Shepherd, A. Comparison of electron microscopic techniques for enumeration of endogenous retrovirus in mouse and Chinese hamster cell lines used for production of biologics. J. Virol. Methods 2003, 108, 91–96. [Google Scholar] [CrossRef]
- Cureton, D.K.; Massol, R.H.; Whelan, S.P.J.; Kirchhausen, T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010, 6, e1001127. [Google Scholar] [CrossRef]
- Wen, K.; Ortmann, A.C.; Suttle, C.A. Accurate estimation of viral abundance by epifluorescence microscopy. Appl. Environ. Microbiol. 2004, 70, 3862–3867. [Google Scholar] [CrossRef]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef]
- Patel, A.; Noble, R.T.; Steele, J.A.; Schwalbach, M.S.; Hewson, I.; Fuhrman, J.A. Virus and prokaryote enumeration from planktonic aquatic environments by epifluorescence microscopy with SYBR Green I. Nat. Protoc. 2007, 2, 269–276. [Google Scholar] [CrossRef]
- Hennes, K.P.; Suttle, C.A. Direct counts of viruses in natural waters and laboratory cultures by epifluorescence microscopy. Limnol. Oceanogr. 1995, 40, 1050–1055. [Google Scholar] [CrossRef]
- Weinbauer, M.G.; Suttle, C.A. Comparison of epifluorescence and transmission electron microscopy for counting viruses in natural marine waters. Aquat. Microb. Ecol. 1997, 13, 225–232. [Google Scholar] [CrossRef]
- Brecher, G.; Schneiderman, M.; Williams, G.Z. Evaluation of electronic red blood cell counter. Am. J. Clin. Pathol. 1956, 26, 1439–1449. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, H.; Chen, Y.; Zhou, Q.; Wang, R.; Xia, B.; Ma, D.; Luo, K.; Liu, Q. Translocation of rigid rod-shaped virus through various solid-state nanopores. Anal. Chem. 2016, 88, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Billinge, E.R.; Muzard, J.; Platt, M. Tunable resistive pulse sensing as a tool to monitor analyte induced particle aggregation. Nanomater. Nanosci. 2013, 1, 1. [Google Scholar] [CrossRef]
- Vogel, R.; Willmott, G.; Kozak, D.; Roberts, G.S.; Anderson, W.; Groenewegen, L.; Glossop, B.; Barnett, A.; Turner, A.; Trau, M. Quantitative sizing of nano/microparticles with a tunable elastomeric pore sensor. Anal. Chem. 2011, 83, 3499–3506. [Google Scholar] [CrossRef]
- Weatherall, E.; Willmott, G.R. Applications of tunable resistive pulse sensing. Analyst 2015, 140, 3318–3334. [Google Scholar] [CrossRef]
- Kozak, D.; Anderson, W.; Vogel, R.; Chen, S.; Antaw, F.; Trau, M. Simultaneous size and ζ-potential measurements of individual nanoparticles in dispersion using size-tunable pore sensors. ACS Nano 2012, 6, 6990–6997. [Google Scholar] [CrossRef]
- Kamentsky, L.A.; Melamed, M.R.; Derman, H. Spectrophotometer: New instrument for ultrarapid cell analysis. Science 1965, 150, 630–631. [Google Scholar] [CrossRef]
- Logan, M.; Manalil, J.; Notte, C.; Kearse, C.; George, S.; Zeiser, A.; Farrell, P.; Aucoin, M.G. A flow cytometric granularity assay for the quantification of infectious virus. Vaccine 2019, 37, 7090–7099. [Google Scholar] [CrossRef]
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. J. Virol. 2018, 92, e01765-17. [Google Scholar] [CrossRef]
- Steen, H.B. Flow cytometer for measurement of the light scattering of viral and other submicroscopic particles. Cytom. Part A J. Int. Soc. Anal. Cytol. 2004, 57, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Hercher, M.; Mueller, W.; Shapiro, H.M. Detection and discrimination of individual viruses by flow cytometry. J. Histochem. Cytochem. 1979, 27, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, C.L.; Kathy, R.F.; Rowlen, K.L. Design and characterization of a compact dual channel virus counter. Cytom. Part A 2005, 65, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zicari, S.; Arakelyan, A.; Fitzgerald, W.; Zaitseva, E.; Chernomordik, L.V.; Margolis, L.; Grivel, J.-C. Evaluation of the maturation of individual Dengue virions with flow virometry. Virology 2016, 488, 20–27. [Google Scholar] [CrossRef]
- Arakelyan, A.; Fitzgerald, W.; Margolis, L.; Grivel, J.-C. Nanoparticle-based flow virometry for the analysis of individual virions. J. Clin. Investig. 2013, 123, 3716–3727. [Google Scholar] [CrossRef]
- Arakelyan, A.; Fitzgerald, W.; Zicari, S.; Vanpouille, C.; Margolis, L. Extracellular vesicles carry HIV Env and facilitate HIV infection of human lymphoid tissue. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Gaudin, R.; Barteneva, N.S. Sorting of small infectious virus particles by flow virometry reveals distinct infectivity profiles. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Baer, A.; Kehn-Hall, K. Viral concentration determination through plaque assays: Using traditional and novel overlay systems. J. Vis. Exp. JoVE 2014, e52065. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Ngunjiri, J.M.; Sekellick, M.J.; Marcus, P.I. Clonogenic assay of type A influenza viruses reveals noninfectious cell-killing (apoptosis-inducing) particles. J. Virol. 2008, 82, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Smither, S.J.; Lear-Rooney, C.; Biggins, J.; Pettitt, J.; Lever, M.S.; Olinger, G.G., Jr. Comparison of the plaque assay and 50% tissue culture infectious dose assay as methods for measuring filovirus infectivity. J. Virol. Methods 2013, 193, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I.; Cieciura, S.J. Clonal growth of mammalian cells in vitro; growth characteristics of colonies from single HeLa cells with and without a feeder layer. J. Exp. Med. 1956, 103, 273–283. [Google Scholar] [CrossRef]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic assay: Adherent cells. J. Vis. Exp. JoVE 2011, 49, e2573. [Google Scholar] [CrossRef]
- Marcus, P.I.; Sekellick, M.J. Cell killing by viruses: I. Comparison of cell-killing, plaque-forming, and defective-interfering particles of vesicular stomatitis virus. Virology 1974, 57, 321–338. [Google Scholar] [CrossRef]
- Von Magnus, P. Incomplete Forms of Influenza Virus. Adv. Virus Res. 1954, 2, 59–79. [Google Scholar]
- Huang, A.S.; Baltimore, D. Defective viral particles and viral disease processes. Nature 1970, 226, 325–327. [Google Scholar] [CrossRef]
- Pattnaik, A.K.; Wertz, G.W. Cells that express all five proteins of vesicular stomatitis virus from cloned cDNAs support replication, assembly, and budding of defective interfering particles. Proc. Natl. Acad. Sci. USA 1991, 88, 1379–1383. [Google Scholar] [CrossRef]
- Calain, P.; Curran, J.; Kolakofsky, D.; Roux, L. Molecular cloning of natural paramyxovirus copy-back defective interfering RNAs and their expression from DNA. Virology 1992, 191, 62–71. [Google Scholar] [CrossRef]
- Lazzarini, R.A.; Keene, J.D.; Schubert, M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 1981, 26 Pt 2, 145–154. [Google Scholar] [CrossRef]
- Albertini, A.M.; Hofer, M.; Calos, M.P.; Miller, J.H. On the formation of spontaneous deletions: The importance of short sequence homologies in the generation of large deletions. Cell 1982, 29, 319–328. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Genoyer, E.; López, C.B. The impact of defective viruses on infection and immunity. Annu. Rev. Virol. 2019, 6, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; López, C.B. Defective viral genomes are key drivers of the virus–host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Molecular basis of genetic variation of viruses: Error-prone replication. In Virus as Populations (Second Edition): Composition, Complexity, Quasispecies, Dynamics, and Biological Implications; Elsevier: Amsterdam, The Netherlands, 2020; pp. 35–71. [Google Scholar]
- Scearce, L.; Pierce, J.; McInroy, B.; Masker, W. Deletion mutagenesis independent of recombination in bacteriophage T7. J. Bacteriol. 1991, 173, 869–878. [Google Scholar] [CrossRef][Green Version]
- Holland, J.J.; Villarreal, L.P.; Breindl, M.; Semler, B.L.; Kohne, D. Defective interfering virus particles attenuate virus lethality in vivo and in vitro. In Animal Virology; Elsevier: Amsterdam, The Netherlands, 1976; pp. 773–786. [Google Scholar]
- Yin, J. Evolution of Bacteriophage T7 in a Growing Plaque. J. Bacteriol. 1993, 175, 1272–1277. [Google Scholar] [CrossRef]
- Knorr, D.; Mullin, R.; Hearne, P.; Morris, T. De novo generation of defective interfering RNAs of tomato bushy stunt virus by high multiplicity passage. Virology 1991, 181, 193–202. [Google Scholar] [CrossRef]
- Kong, D.; Yin, J. Whole-virus Vaccine Development by Continuous Culture on a Complementing Host. Bio/Technology 1995, 13, 583–586. [Google Scholar] [CrossRef]
- White, K.A.; Morris, T.J. Nonhomologous RNA recombination in tombusviruses: Generation and evolution of defective interfering RNAs by stepwise deletions. J. Virol. 1994, 68, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Russell, A.B. Library-based analysis reveals segment and length dependent characteristics of defective influenza genomes. PLoS Pathog. 2021, 17, e1010125. [Google Scholar] [CrossRef] [PubMed]
- Duhaut, S.; McCauley, J. Defective RNAs inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology 1996, 216, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Van der Most, R.; Bredenbeek, P.; Spaan, W. A domain at the 3′end of the polymerase gene is essential for encapsidation of coronavirus defective interfering RNAs. J. Virol. 1991, 65, 3219–3226. [Google Scholar] [CrossRef]
- Wu, C.; Harper, L.; Ben-Porat, T. Molecular basis for interference of defective interfering particles of pseudorabies virus with replication of standard virus. J. Virol. 1986, 59, 308–317. [Google Scholar] [CrossRef]
- Yawei, N.; Kemp, M.C. Subgenomic S1 segments are packaged by avian reovirus defective interfering particles having an S1 segment deletion. Virus Res. 1994, 32, 329–342. [Google Scholar] [CrossRef]
- Belicha-Villanueva, A.; Rodriguez-Madoz, J.R.; Maamary, J.; Baum, A.; Bernal-Rubio, D.; Minguito de la Escalera, M.; Fernandez-Sesma, A.; García-Sastre, A. Recombinant influenza A viruses with enhanced levels of PB1 and PA viral protein expression. J. Virol. 2012, 86, 5926–5930. [Google Scholar] [CrossRef]
- Kupke, S.Y.; Riedel, D.; Frensing, T.; Zmora, P.; Reichl, U. A novel type of influenza A virus-derived defective interfering particle with nucleotide substitutions in its genome. J. Virol. 2019, 93, e01786-18. [Google Scholar] [CrossRef]
- Genoyer, E.; López, C.B. Defective viral genomes alter how Sendai virus interacts with cellular trafficking machinery, leading to heterogeneity in the production of viral particles among infected cells. J. Virol. 2019, 93, e01579-18. [Google Scholar] [CrossRef]
- Horodyski, F.M.; Nichol, S.T.; Spindler, K.R.; Holland, J.J. Properties of DI particle resistant mutants of vesicular stomatitis virus isolated from persistent infections and from undiluted passages. Cell 1983, 33, 801–810. [Google Scholar] [CrossRef]
- DePolo, N.J.; Giachetti, C.; Holland, J.J. Continuing coevolution of virus and defective interfering particles and of viral genome sequences during undiluted passages: Virus mutants exhibiting nearly complete resistance to formerly dominant defective interfering particles. J. Virol. 1987, 61, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Matsumoto, S. Interfering and noninterfering defective particles generated by a rabies small plaque variant virus. Virology 1977, 76, 60–71. [Google Scholar] [CrossRef]
- Jacobson, S.; Pfau, C.J. Viral pathogenesis and resistance to defective interfering particles. Nature 1980, 283, 311–313. [Google Scholar] [CrossRef]
- Weiss, B.; Schlesinger, S. Defective interfering particles of Sindbis virus do not interfere with the homologous virus obtained from persistently infected BHK cells but do interfere with Semliki Forest virus. J. Virol. 1981, 37, 840–844. [Google Scholar] [CrossRef]
- Brinton, M.A.; Fernandez, A.V. A replication-efficient mutant of West Nile virus is insensitive to DI particle interference. Virology 1983, 129, 107–115. [Google Scholar] [CrossRef]
- Garcia-Arriaza, J.; Manrubia, S.C.; Toja, M.; Domingo, E.; Escarmis, C. Evolutionary transition toward defective RNAs that are infectious by complementation. J. Virol. 2004, 78, 11678–11685. [Google Scholar] [CrossRef]
- Lucía-Sanz, A.; Aguirre, J.; Manrubia, S. Theoretical approaches to disclosing the emergence and adaptive advantages of multipartite viruses. Curr. Opin. Virol. 2018, 33, 89–95. [Google Scholar] [CrossRef]
- White, K.A.; Morris, T.J. Recombination between defective tombusvirus RNAs generates functional hybrid genomes. Proc. Natl. Acad. Sci. USA 1994, 91, 3642–3646. [Google Scholar] [CrossRef]
- Raju, R.; Subramaniam, S.; Hajjou, M. Genesis of Sindbis virus by in vivo recombination of nonreplicative RNA precursors. J. Virol. 1995, 69, 7391–7401. [Google Scholar] [CrossRef]
- Koonin, E.V. Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150442. [Google Scholar] [CrossRef] [PubMed]
- Bellett, A.J.; Cooper, P.D. Some properties of the transmissible interfering component of vesicular stomatitis virus preparations. J. Gen. Microbiol. 1959, 21, 498–509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Szathmary, E. Natural selection and dynamical coexistence of defective and complementing virus segments. J. Theor. Biol. 1992, 157, 383–406. [Google Scholar] [CrossRef]
- Kirkwood, T.B.; Bangham, C.R. Cycles, chaos, and evolution in virus cultures: A model of defective interfering particles. Proc. Natl. Acad. Sci. USA 1994, 91, 8685–8689. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.A. Within-host spatial dynamics of viruses and defective interfering particles. J. Theor. Biol. 2000, 206, 279–290. [Google Scholar] [CrossRef]
- Akkina, R.K.; Chambers, T.M.; Nayak, D.P. Mechanism of interference by defective-interfering particles of influenza virus: Differential reduction of intracellular synthesis of specific polymerase proteins. Virus Res. 1984, 1, 687–702. [Google Scholar] [CrossRef]
- Khan, S.R.; Lazzarini, R.A. The relationship between autointerference and the replication of defective interfering particle. Virology 1977, 77, 189–201. [Google Scholar] [CrossRef]
- Stauffer Thompson, K.A.; Rempala, G.A.; Yin, J. Multiple-hit inhibition of infection by defective interfering particles. J. Gen. Virol. 2009, 90 Pt 4, 888–899. [Google Scholar] [CrossRef]
- Sekellick, M.J.; Marcus, P.I. Viral interference by defective particles of vesicular stomatitis virus measured in individual cells. Virology 1980, 104, 247–252. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Schaffer, D.V.; Arkin, A.P. Theoretical design of a gene therapy to prevent AIDS but not human immunodeficiency virus type 1 infection. J. Virol. 2003, 77, 10028–10036. [Google Scholar] [CrossRef]
- Harty, R.N.; Caughman, G.B.; Holden, V.R.; O’Callaghan, D.J. Characterization of the myristylated polypeptide encoded by the UL1 gene that is conserved in the genome of defective interfering particles of equine herpesvirus 1. J. Virol. 1993, 67, 4122–4132. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.; Saunders, K.; Pinner, M.S.; Wong, S.M. Novel Defective Interfering DNAs Associated with Ageratum Yellow Vein Geminivirus Infection ofAgeratum conyzoides. Virology 1997, 239, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Bay, P.; Reichmann, M. UV inactivation of the biological activity of defective interfering particles generated by vesicular stomatitis virus. J. Virol. 1979, 32, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.M.; Simon, E.H. Defective interfering particles in monolayer-propagated Newcastle disease virus. Virology 1976, 69, 298–303. [Google Scholar] [CrossRef]
- Rima, B.; Davidson, W.; Martin, S. The role of defective interfering particles in persistent infection of Vero cells by measles virus. J. Gen. Virol. 1977, 35, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.P. Defective interfering influenza viruses. Annu. Rev. Microbiol. 1980, 34, 619–644. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Fujiwara, K.; Lai, M.M. Defective interfering particles of coronavirus. In Coronaviruses; Springer: New York, NY, USA, 1987; pp. 187–195. [Google Scholar]
- Nomburg, J.; Meyerson, M.; DeCaprio, J.A. Pervasive generation of non-canonical subgenomic RNAs by SARS-CoV-2. Genome Med. 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Manzoni, T.B.; López, C.B. Defective (interfering) viral genomes re-explored: Impact on antiviral immunity and virus persistence. Future Virol. 2018, 13, 493–503. [Google Scholar] [CrossRef]
- Aaskov, J.; Buzacott, K.; Thu, H.M.; Lowry, K.; Holmes, E.C. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 2006, 311, 236–238. [Google Scholar] [CrossRef]
- Li, D.; Lott, W.B.; Lowry, K.; Jones, A.; Thu, H.M.; Aaskov, J. Defective interfering viral particles in acute dengue infections. PLoS ONE 2011, 6, e19447. [Google Scholar] [CrossRef]
- Wang, S.; Chan, K.W.; Naripogu, K.B.; Swarbrick, C.M.; Aaskov, J.; Vasudevan, S.G. Subgenomic RNA from dengue virus type 2 suppresses replication of dengue virus genomes and interacts with virus-encoded NS3 and NS5 proteins. ACS Infect. Dis. 2020, 6, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Saira, K.; Lin, X.; DePasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J. Virol. 2013, 87, 8064–8074. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.P.; Chambers, T.M.; Akkina, R.K. Defective-interfering (DI) RNAs of influenza viruses: Origin, structure, expression, and interference. Curr. Top. Microbiol. Immunol. 1985, 114, 103–151. [Google Scholar] [PubMed]
- Jennings, P.A.; Finch, J.T.; Winter, G.; Robertson, J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 1983, 34, 619–627. [Google Scholar] [CrossRef]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Sun, Y.; Jain, D.; Koziol-White, C.J.; Genoyer, E.; Gilbert, M.; Tapia, K.; Panettieri Jr, R.A.; Hodinka, R.L.; López, C.B. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog. 2015, 11, e1005122. [Google Scholar] [CrossRef]
- Xie, Q.; Cao, Y.; Su, J.; Wu, J.; Wu, X.; Wan, C.; He, M.; Ke, C.; Zhang, B.; Zhao, W. Two deletion variants of Middle East respiratory syndrome coronavirus found in a patient with characteristic symptoms. Arch. Virol. 2017, 162, 2445–2449. [Google Scholar] [CrossRef]
- Lu, X.; Rowe, L.A.; Frace, M.; Stevens, J.; Abedi, G.R.; Elnile, O.; Banassir, T.; Al-Masri, M.; Watson, J.T.; Assiri, A. Spike gene deletion quasispecies in serum of patient with acute MERS-CoV infection. J. Med. Virol. 2017, 89, 542–545. [Google Scholar] [CrossRef]
- Wong, C.H.; Ngan, C.Y.; Goldfeder, R.L.; Idol, J.; Kuhlberg, C.; Maurya, R.; Kelly, K.; Omerza, G.; Renzette, N.; De Abreu, F. Reduced subgenomic RNA expression is a molecular indicator of asymptomatic SARS-CoV-2 infection. Commun. Med. 2021, 1, 1–12. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Yang, Y.; Lyu, T.; Zhou, R.; He, X.; Ye, K.; Xie, Q.; Zhu, L.; Chen, T.; Shen, C.; Wu, Q. The antiviral and antitumor effects of defective interfering particles/genomes and their mechanisms. Front. Microbiol. 2019, 10, 1852. [Google Scholar] [CrossRef]
- Marcus, P.I.; Rojek, J.M.; Sekellick, M.J. Interferon induction and/or production and its suppression by influenza A viruses. J. Virol. 2005, 79, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Sekellick, M.J.; Marcus, P.I. Interferon induction by viruses. XIV. Development of interferon inducibility and its inhibition in chick embryo cells “aged” in vitro. J. Interferon Res. 1985, 5, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Marcus, P.I. Interferon induction: Dose-response curves. Methods Enzymol. 1986, 119, 106–114. [Google Scholar]
- Marcus, P.I.; Sekellick, M.J. Defective interfering particles with covalently linked [±] RNA induce interferon. Nature 1977, 266, 815–819. [Google Scholar] [CrossRef]
- Carter, W.A.; De Clercq, E. Viral infection and host defense. Science 1974, 186, 1172–1178. [Google Scholar] [CrossRef]
- García-Sastre, A. Mechanisms of inhibition of the host interferon α/β-mediated antiviral responses by viruses. Microbes Infect. 2002, 4, 647–655. [Google Scholar] [CrossRef]
- Krug, R.M.; Yuan, W.; Noah, D.L.; Latham, A.G. Intracellular warfare between human influenza viruses and human cells: The roles of the viral NS1 protein. Virology 2003, 309, 181–189. [Google Scholar] [CrossRef]
- Felt, S.A.; Sun, Y.; Jozwik, A.; Paras, A.; Habibi, M.S.; Nickle, D.; Anderson, L.; Achouri, E.; Feemster, K.A.; Cárdenas, A.M. Detection of respiratory syncytial virus defective genomes in nasal secretions is associated with distinct clinical outcomes. Nat. Microbiol. 2021, 6, 672–681. [Google Scholar] [CrossRef]
- López, C.B.; Yount, J.S.; Hermesh, T.; Moran, T.M. Sendai virus infection induces efficient adaptive immunity independently of type I interferons. J. Virol. 2006, 80, 4538–4545. [Google Scholar] [CrossRef] [PubMed]
- Yount, J.S.; Gitlin, L.; Moran, T.M.; Lopez, C.B. MDA5 participates in the detection of paramyxovirus infection and is essential for the early activation of dendritic cells in response to Sendai Virus defective interfering particles. J. Immunol. 2008, 180, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gil, L.; Goff, P.; Hai, R.; Garcia-Sastre, A.; Shaw, M.; Palese, P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J. Virol. 2013, 87, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Vasou, A.; Sultanoglu, N.; Goodbourn, S.; Randall, R.E.; Kostrikis, L.G. Targeting pattern recognition receptors (PRR) for vaccine adjuvantation: From synthetic PRR agonists to the potential of defective interfering particles of viruses. Viruses 2017, 9, 186. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Dove, B.K.; Meng, B.; Scott, P.D.; Taylor, I.; Cheung, L.; Hallis, B.; Marriott, A.C.; Carroll, M.W.; Easton, A.J. Comparison of the protection of ferrets against pandemic 2009 influenza A virus (H1N1) by 244 DI influenza virus and oseltamivir. Antivir. Res. 2012, 96, 376–385. [Google Scholar] [CrossRef]
- Harding, A.T.; Haas, G.D.; Chambers, B.S.; Heaton, N.S. Influenza viruses that require 10 genomic segments as antiviral therapeutics. PLoS Pathog. 2019, 15, e1008098. [Google Scholar] [CrossRef]
- Klimatcheva, E.; Planelles, V.; Day, S.L.; Fulreader, F.; Renda, M.J.; Rosenblatt, J. Defective lentiviral vectors are efficiently trafficked by HIV-1 and inhibit its replication. Mol. Ther. 2001, 3, 928–939. [Google Scholar] [CrossRef]
- An, D.S.; Morizono, K.; Li, Q.-X.; Mao, S.H.; Lu, S.; Chen, I.S. An inducible human immunodeficiency virus type 1 (HIV-1) vector which effectively suppresses HIV-1 replication. J. Virol. 1999, 73, 7671–7677. [Google Scholar] [CrossRef]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.-R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef]
- Tanner, E.J.; Kirkegaard, K.A.; Weinberger, L.S. Exploiting genetic interference for antiviral therapy. PLoS Genet. 2016, 12, e1005986. [Google Scholar] [CrossRef]
- Metzger, V.T.; Lloyd-Smith, J.O.; Weinberger, L.S. Autonomous targeting of infectious superspreaders using engineered transmissible therapies. PLoS Comput. Biol. 2011, 7, e1002015. [Google Scholar] [CrossRef] [PubMed]
- Notton, T.; Sardanyés, J.; Weinberger, A.D.; Weinberger, L.S. The case for transmissible antivirals to control population-wide infectious disease. Trends Biotechnol. 2014, 32, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lin, M.-H.; Rawle, D.J.; Jin, H.; Wu, Z.; Wang, L.; Lor, M.; Hussain, M.; Aaskov, J.; Harrich, D. Dengue virus-free defective interfering particles have potent and broad anti-dengue virus activity. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rezelj, V.V.; Carrau, L.; Merwaiss, F.; Levi, L.I.; Erazo, D.; Tran, Q.D.; Henrion-Lacritick, A.; Gausson, V.; Suzuki, Y.; Shengjuler, D. Defective viral genomes as therapeutic interfering particles against flavivirus infection in mammalian and mosquito hosts. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Makino, S.; Fujioka, N.; Fujiwara, K. Structure of the intracellular defective viral RNAs of defective interfering particles of mouse hepatitis virus. J. Virol. 1985, 54, 329–336. [Google Scholar] [CrossRef]
- Yao, S.; Narayanan, A.; Majowicz, S.A.; Jose, J.; Archetti, M. A synthetic defective interfering SARS-CoV-2. PeerJ 2021, 9, e11686. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Vasen, G.; Pablo, M.; Chen, X.; Beutler, N.; Kumar, A.; Tanner, E.; Illouz, S.; Rahgoshay, D.; Burnett, J. Identification of a therapeutic interfering particle—A single-dose SARS-CoV-2 antiviral intervention with a high barrier to resistance. Cell 2021, 184, 6022–6036. [Google Scholar] [CrossRef]
- Benn, C.S.; Fisker, A.B.; Rieckmann, A.; Sørup, S.; Aaby, P. Vaccinology: Time to change the paradigm? Lancet Infect. Dis. 2020, 20, e274–e283. [Google Scholar] [CrossRef]
- Xiao, Y.; Lidsky, P.V.; Shirogane, Y.; Aviner, R.; Wu, C.-T.; Li, W.; Zheng, W.; Talbot, D.; Catching, A.; Doitsh, G. A defective viral genome strategy elicits broad protective immunity against respiratory viruses. Cell 2021, 184, 6037–6051. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef]
- Brooke, C.B.; Ince, W.L.; Wrammert, J.; Ahmed, R.; Wilson, P.C.; Bennink, J.R.; Yewdell, J.W. Most influenza a virions fail to express at least one essential viral protein. J. Virol. 2013, 87, 3155–3162. [Google Scholar] [CrossRef]
- Diefenbacher, M.; Sun, J.; Brooke, C.B. The parts are greater than the whole: The role of semi-infectious particles in influenza A virus biology. Curr. Opin. Virol. 2018, 33, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-Y.; Vafabakhsh, R.; Doğanay, S.; Gao, Q.; Ha, T.; Palese, P. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc. Natl. Acad. Sci. USA 2012, 109, 9101–9106. [Google Scholar] [CrossRef]
- Timm, A.C.; Warrick, J.W.; Yin, J. Quantitative profiling of innate immune activation by viral infection in single cells. Integr. Biol. 2017, 9, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.W.; Timm, A.; Swick, A.; Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE 2016, 11, e0145081. [Google Scholar] [CrossRef]
- Zhu, Y.; Yongky, A.; Yin, J. Growth of an RNA virus in single cells reveals a broad fitness distribution. Virology 2009, 385, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, F.; Timm, A.; Yin, J. High-Throughput Single-Cell Kinetics of Virus Infections in the Presence of Defective Interfering Particles. J. Virol. 2015, 90, 1599–1612. [Google Scholar] [CrossRef]
- Stiefel, P.; Schmidt, F.I.; Dörig, P.; Behr, P.; Zambelli, T.; Vorholt, J.A.; Mercer, J. Cooperative vaccinia infection demonstrated at the single-cell level using FluidFM. Nano Lett. 2012, 12, 4219–4227. [Google Scholar] [CrossRef]
- Delbrück, M. The burst size distribution in the growth of bacterial viruses (bacteriophages). J. Bacteriol. 1945, 50, 131–135. [Google Scholar] [CrossRef]
- Schulte, M.B.; Andino, R. Single-cell analysis uncovers extensive biological noise in poliovirus replication. J. Virol. 2014, 88, 6205–6212. [Google Scholar] [CrossRef]
- Heldt, F.S.; Kupke, S.Y.; Dorl, S.; Reichl, U.; Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 2015, 6, 8938. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.; Ross, J.; McAdams, H.H. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics 1998, 149, 1633–1648. [Google Scholar] [CrossRef] [PubMed]
- Hensel, S.C.; Rawlings, J.B.; Yin, J. Stochastic Kinetic Modeling of Vesicular Stomatitis Virus Intracellular Growth. Bull. Math. Biol. 2009, 71, 1671–1692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srivastava, R.; You, L.; Summers, J.; Yin, J. Stochastic versus Deterministic Modeling of Intracellular Viral Kinetics. J. Theor. Biol. 2002, 218, 309–321. [Google Scholar] [CrossRef]
- Schelker, M.; Mair, C.M.; Jolmes, F.; Welke, R.-W.; Klipp, E.; Herrmann, A.; Flöttmann, M.; Sieben, C. Viral RNA degradation and diffusion act as a bottleneck for the influenza A virus infection efficiency. PLoS Comput. Biol. 2016, 12, e1005075. [Google Scholar] [CrossRef]
- Jacobs, N.T.; Onuoha, N.O.; Antia, A.; Steel, J.; Antia, R.; Lowen, A.C. Incomplete influenza A virus genomes occur frequently but are readily complemented during localized viral spread. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Phipps, K.L.; Ganti, K.; Jacobs, N.T.; Lee, C.-Y.; Carnaccini, S.; White, M.C.; Manandhar, M.; Pickett, B.E.; Tan, G.S.; Ferreri, L.M. Collective interactions augment influenza A virus replication in a host-dependent manner. Nat. Microbiol. 2020, 5, 1158–1169. [Google Scholar] [CrossRef]
- Nakatsu, S.; Sagara, H.; Sakai-Tagawa, Y.; Sugaya, N.; Noda, T.; Kawaoka, Y. Complete and incomplete genome packaging of influenza A and B viruses. mBio 2016, 7, e01248-16. [Google Scholar] [CrossRef]
- Sun, J.; Vera, J.C.; Drnevich, J.; Lin, Y.T.; Ke, R.; Brooke, C.B. Single cell heterogeneity in influenza A virus gene expression shapes the innate antiviral response to infection. PLoS Pathog. 2020, 16, e1008671. [Google Scholar] [CrossRef]
- Pauly, M.D.; Procario, M.C.; Lauring, A.S. A novel twelve class fluctuation test reveals higher than expected mutation rates for influenza A viruses. eLife 2017, 6, e26437. [Google Scholar] [CrossRef]
- Pérez-Cidoncha, M.; Killip, M.J.; Oliveros, J.C.; Asensio, V.J.; Fernández, Y.; Bengoechea, J.A.; Randall, R.E.; Ortín, J. An unbiased genetic screen reveals the polygenic nature of the influenza virus anti-interferon response. J. Virol. 2014, 88, 4632–4646. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef]
- Marcus, P.I.; Ngunjiri, J.M.; Sekellick, M.J. Dynamics of biologically active subpopulations of influenza virus: Plaque-forming, noninfectious cell-killing, and defective interfering particles. J. Virol. 2009, 83, 8122–8130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herrera, M.; Garcia-Arriaza, J.; Pariente, N.; Escarmis, C.; Domingo, E. Molecular Basis for a Lack of Correlation between Viral Fitness and Cell Killing Capacity. PLoS Pathog. 2007, 3, e53. [Google Scholar] [CrossRef] [PubMed]
- Furió, V.; Garijo, R.; Durán, M.; Moya, A.; Bell, J.C.; Sanjuán, R. Relationship between within-host fitness and virulence in the vesicular stomatitis virus: Correlation with partial decoupling. J. Virol. 2012, 86, 12228–12236. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, P.; de la Iglesia, F.; Elena, S.F. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco etch virus. J. Virol. 2007, 81, 12979–12984. [Google Scholar] [CrossRef]
- Van den Pol, A.N.; Davis, J.N. Highly attenuated recombinant vesicular stomatitis virus VSV-12′ GFP displays immunogenic and oncolytic activity. J. Virol. 2013, 87, 1019–1034. [Google Scholar] [CrossRef]
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895. [Google Scholar] [CrossRef]
- McCormick, W.; Mermel, L.A. The basic reproductive number and particle-to-plaque ratio: Comparison of these two parameters of viral infectivity. Virol. J. 2021, 18, 1–4. [Google Scholar] [CrossRef]
- Addetia, A.; Phung, Q.; Bradley, B.T.; Lin, M.J.; Zhu, H.; Xie, H.; Huang, M.-L.; Greninger, A.L. In vivo generation of BK and JC polyomavirus defective viral genomes in human urine samples associated with higher viral loads. J. Virol. 2021, 95, e00250-21. [Google Scholar] [CrossRef]
- Baltes, A.; Akpinar, F.; Inankur, B.; Yin, J. Inhibition of infection spread by co-transmitted defective interfering particles. PLoS ONE 2017, 12, e0184029. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, F.; Inankur, B.; Yin, J. S patial-Temporal Patterns of Viral Amplification and Interference Initiated by a Single Infected Cell. J. Virol. 2016, 90, 7552–7566. [Google Scholar] [CrossRef] [PubMed]
- Kupke, S.Y.; Ly, L.-H.; Börno, S.T.; Ruff, A.; Timmermann, B.; Vingron, M.; Haas, S.; Reichl, U. Single-Cell Analysis Uncovers a Vast Diversity in Intracellular Viral Defective Interfering RNA Content Affecting the Large Cell-to-Cell Heterogeneity in Influenza A Virus Replication. Viruses 2020, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. Molecular determinants of the ratio of inert to infectious virus particles. Prog. Mol. Biol. Transl. Sci. 2015, 129, 285–326. [Google Scholar] [PubMed]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef]
- Mosca, J.; Pitha, P. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 1986, 6, 2279–2283. [Google Scholar]
- Kramer, M.J.; Dennin, R.; Kramer, C.; Jones, G.; Connell, E.; Rolon, N.; Gruarin, A.; Kale, R.; Trown, P.W. Cell and virus sensitivity studies with recombinant human alpha interferons. J. Interferon Res. 1983, 3, 425–435. [Google Scholar] [CrossRef]
- Andzhaparidze, O.G.; Bogomolova, N.N.; Boriskin, Y.S.; Bektemirova, M.S.; Drynov, I.D. Comparative study of rabies virus persistence in human and hamster cell lines. J. Virol. 1981, 37, 1–6. [Google Scholar] [CrossRef]
- Rausell, A.; Muñoz, M.; Martinez, R.; Roger, T.; Telenti, A.; Ciuffi, A. Innate immune defects in HIV permissive cell lines. Retrovirology 2016, 13, 1–10. [Google Scholar] [CrossRef]
- Petricoin, E., 3rd; David, M.; Fang, H.; Grimley, P.; Larner, A.C.; Vande Pol, S. Human cancer cell lines express a negative transcriptional regulator of the interferon regulatory factor family of DNA binding proteins. Mol. Cell. Biol. 1994, 14, 1477–1486. [Google Scholar]
- Amineva, S.P.; Aminev, A.G.; Gern, J.E.; Palmenberg, A.C. Comparison of rhinovirus A infection in human primary epithelial and HeLa cells. J. Gen. Virol. 2011, 92 Pt 11, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Brockman-Schneider, R.; Bochkov, Y.A.; Pasic, T.R.; Gern, J.E. Biological characteristics and propagation of human rhinovirus-C in differentiated sinus epithelial cells. Virology 2013, 436, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; Ploss, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Huang, S.X.; de Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Detection Type | Counting | Throughput | Detection Limit | References |
|---|---|---|---|---|---|
| Transmission electron microscopy | Physical virus particle | Manual and automated | 100 particles per grid | ≥107 particles/mL | Borries (1938) Timm (2014) Blancett (2017) Roingeard (2019) |
| Epifluorescence microscopy | Fluorophore-labelled virus particle | Manual and automated | 100 particles per grid | ≥107 particles/mL | Chen (2001) Ortmann and Suttle (2009) Parveen (2018) |
| Tunable resistive pulse sensing | Physical virus particle | Automated | 10,000 particles per sec | 107–1010 particles/mL | Akpinar (2015) Yang (2016) |
| Flow virometry | Fluorophore-labelled virus particle | Automated | 2000–6000 particles per sec | 105–109 particles/mL | Rossi (2015) Zamora (2017) |
| Technique | Detection Type | Counting | Time | Units | References |
|---|---|---|---|---|---|
| Plaque assay | Infectious virus particle | Manual | 2–14 days | pfu/mL | Baer and Kehn-Hall (2014) |
| End-point Dilution assay | Infectious virus titer for 50% CPE | Manual | Varies depending on infection time of virus | TCID50/mL | Flint et al. (2004) Reed and Muench (1938) |
| Clonogenic assay | Cell-killing particle | Manual or Automated | Based on incubation time for visible colonies (1–3 weeks for eukaryotes) | CKPs/mL | Ngunjiri et al. (2008) Franken et al. (2006) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhat, T.; Cao, A.; Yin, J. Virus-like Particles: Measures and Biological Functions. Viruses 2022, 14, 383. https://doi.org/10.3390/v14020383
Bhat T, Cao A, Yin J. Virus-like Particles: Measures and Biological Functions. Viruses. 2022; 14(2):383. https://doi.org/10.3390/v14020383
Chicago/Turabian StyleBhat, Tara, Amy Cao, and John Yin. 2022. "Virus-like Particles: Measures and Biological Functions" Viruses 14, no. 2: 383. https://doi.org/10.3390/v14020383
APA StyleBhat, T., Cao, A., & Yin, J. (2022). Virus-like Particles: Measures and Biological Functions. Viruses, 14(2), 383. https://doi.org/10.3390/v14020383