
Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. CRISPR/Cas9 Plasmid and gRNA Design
2.3. Preparation of AAV
2.4. Quantification of Viral Genomes
2.5. Generation of Recombinant VZV
2.6. Virus Derivation and Growth Curves
2.7. Neuron Cultures
2.8. Fluorescent Microscopy
2.9. Flow Cytometry and Statistical Analyses
3. Results
3.1. Characterization of VZV-Specific gRNAs for In Vitro Specificity
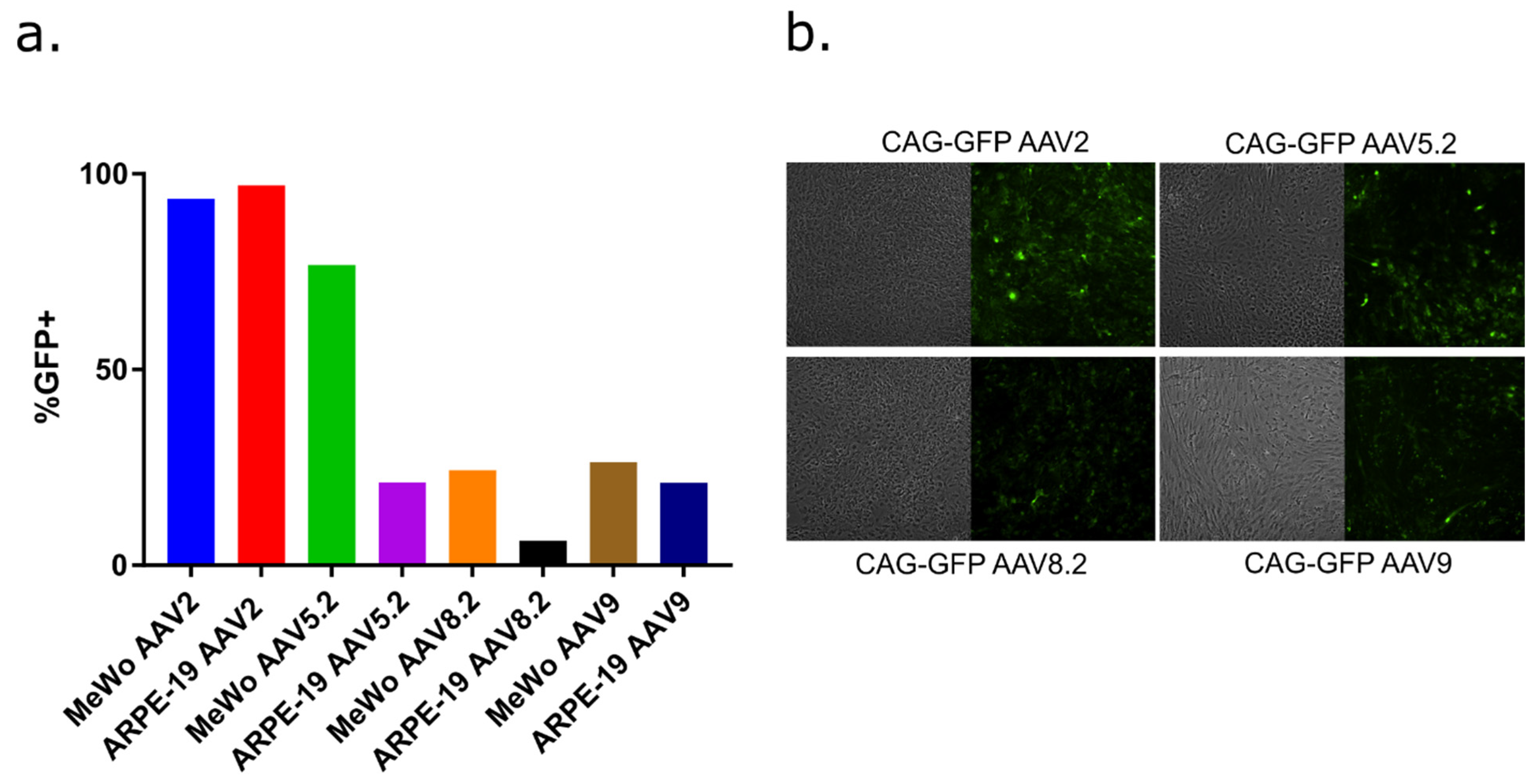
3.2. AAV Serotypes for Delivery to VZV Permissive Epithelial Cells and Human Stem Cell-Derived Neurons
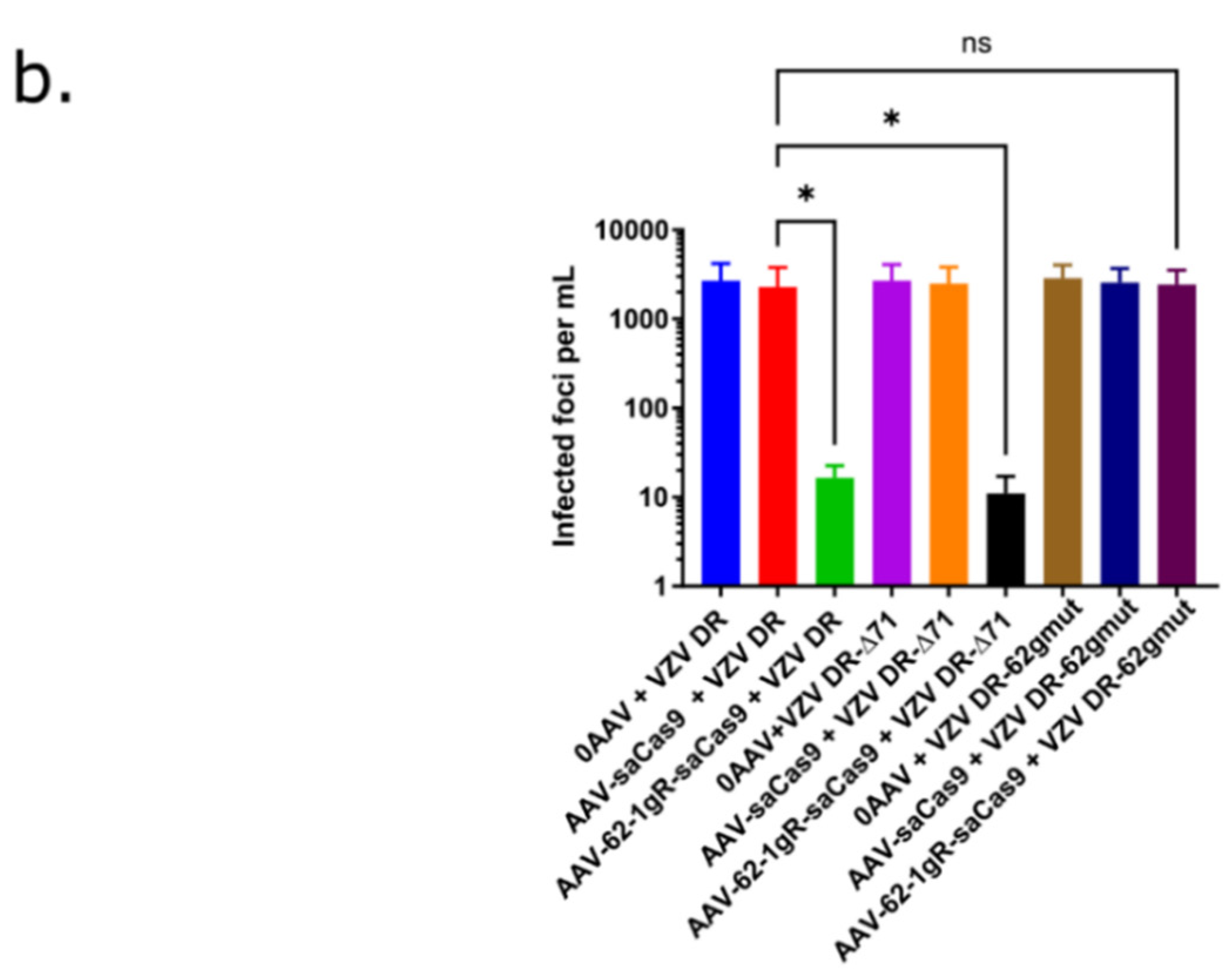
3.3. AAV-62-1gR-saCas9 Reduces VZV Lytic Replication in Epithelial Cells
3.4. AAV-62-1gR-saCas9 Reduces VZV Lytic Replication in hESC-Derived Human Neuron Cultures
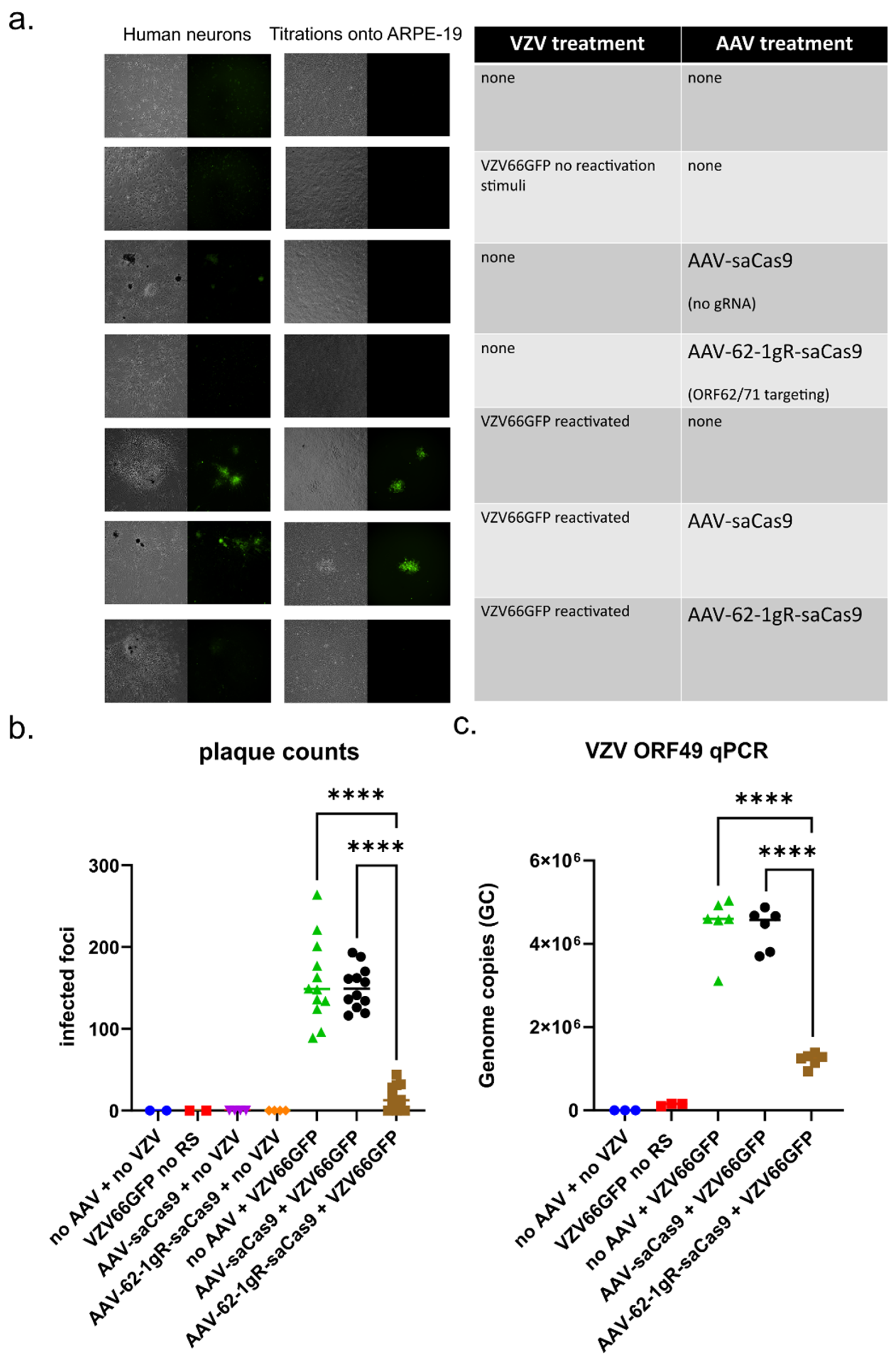
3.5. AAV-62-1gR-saCas9 Reduces VZV Replication Following Reactivation in Model Latently Infected Neuron Cultures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef]
- Johnson, R.W. Herpes Zoster and Postherpetic Neuralgia. Expert Rev. Vaccines 2014, 9, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Gershon, M.D. Pathogenesis and current approaches to control of varicella-zoster virus infections. Clin. Microbiol. Rev. 2013, 26, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Edell, A.R.P.; Cohen, E.J. Herpes Simplex and Herpes Zoster Eye Disease. Eye Contact Lens Sci. Clin. Pract. 2013, 39, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Pavan-Langston, D. Herpes Zoster. Antivirals and Pain Management. Ophthalmology 2008, 115, 13–20. [Google Scholar] [CrossRef]
- Oxman, M.N.; Levin, M.J.; Johnson, G.R.; Schmader, K.E.; Straus, S.E.; Gelb, L.D.; Arbeit, R.D.; Simberkoff, M.S.; Gershon, A.A.; Davis, L.E.; et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 2005, 352, 2271–2284. [Google Scholar] [CrossRef]
- Chun, C.; Weinmann, S.; Riedlinger, K.; Mullooly, J.P.; Houston, H.; Schmid, D.S.; Seward, J.F. Laboratory Characteristics of Suspected Herpes Zoster in Vaccinated Children. Pediatr. Infect. Dis. J. 2011, 30, 721–723. [Google Scholar] [CrossRef]
- Tseng, H.F.; Schmid, D.S.; Harpaz, R.; LaRussa, P.; Jensen, N.J.; Rivailler, P.; Radford, K.; Folster, J.; Jacobsen, S.J. Herpes zoster caused by vaccine-strain varicella zoster virus in an immunocompetent recipient of zoster vaccine. Clin. Infect. Dis. 2014, 58, 1125–1128. [Google Scholar] [CrossRef]
- Lal, H.; Cunningham, A.L.; Godeaux, O.; Chlibek, R.; Diez-Domingo, J.; Hwang, S.-J.; Levin, M.J.; McElhaney, J.E.; Poder, A.; Puig-Barberà, J.; et al. Efficacy of an Adjuvanted Herpes Zoster Subunit Vaccine in Older Adults. N. Engl. J. Med. 2015, 372, 2087–2096. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Lal, H.; Kovac, M.; Chlibek, R.; Hwang, S.-J.; Díez-Domingo, J.; Godeaux, O.; Levin, M.J.; McElhaney, J.E.; Puig-Barberà, J.; et al. Efficacy of the Herpes Zoster Subunit Vaccine in Adults 70 Years of Age or Older. N. Engl. J. Med. 2016, 375, 1019–1032. [Google Scholar] [CrossRef]
- Lal, H.; Poder, A.; Campora, L.; Geeraerts, B.; Oostvogels, L.; Vanden Abeele, C.; Heineman, T.C. Immunogenicity, reactogenicity and safety of 2 doses of an adjuvanted herpes zoster subunit vaccine administered 2, 6 or 12 months apart in older adults: Results of a phase III, randomized, open-label, multicenter study. Vaccine 2018, 36, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Koshy, E.; Mengting, L.; Kumar, H.; Jianbo, W. Epidemiology, treatment and prevention of herpes zoster: A comprehensive review. Indian J. Dermatol. Venereol. Leprol. 2018, 84, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sauerbrei, A. Diagnosis, antiviral therapy, and prophylaxis of varicella-zoster virus infections. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Guedon, J.M.G.; Yee, M.B.; Zhang, M.; Harvey, S.A.K.; Goins, W.F.; Kinchington, P.R. Neuronal changes induced by Varicella Zoster Virus in a rat model of postherpetic neuralgia. Virology 2015, 482, 167–180. [Google Scholar] [CrossRef]
- Sato, H.; Pesnicak, L.; Cohen, J.I. Varicella-zoster virus ORF47 protein kinase, which is required for replication in human T cells, and ORF66 protein kinase, which is expressed during latency, are dispensable for establishment of latency. J. Virol. 2003, 77, 11180–11185. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Badani, H.; Baird, N.L.; White, T.M.; Sanford, B.; Gilden, D. Induction of varicella zoster virus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2017, 23, 152. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Choe, A.; Nagel, M.A.; Gilden, D.; Osterhaus, A.D.M.E.; Cohrs, R.J.; Verjans, G.M.G.M. Restricted Varicella-Zoster Virus Transcription in Human Trigeminal Ganglia Obtained Soon after Death. J. Virol. 2012, 86, 10203–10206. [Google Scholar] [CrossRef]
- Rowe, J.; Greenblatt, R.J.; Liu, D.; Moffat, J.F. Compounds that target host cell proteins prevent varicella-zoster virus replication in culture, ex vivo, and in SCID-Hu mice. Antivir. Res. 2010, 86, 276–285. [Google Scholar] [CrossRef][Green Version]
- Zerboni, L.; Arvin, A. Neuronal Subtype and Satellite Cell Tropism Are Determinants of Varicella-Zoster Virus Virulence in Human Dorsal Root Ganglia Xenografts In Vivo. PLoS Pathog. 2015, 11, e1004989. [Google Scholar] [CrossRef]
- De, C.; Liu, D.; Zheng, B.; Singh, U.S.; Chavre, S.; White, C.; Arnold, R.D.; Hagen, F.K.; Chu, C.K.; Moffat, J.F. β-l-1-[5-(E-2-bromovinyl)-2-(hydroxymethyl)-1,3-(dioxolan-4-yl)] uracil (l-BHDU) prevents varicella-zoster virus replication in a SCID-Hu mouse model and does not interfere with 5-fluorouracil catabolism. Antivir. Res. 2014, 110, 10–19. [Google Scholar] [CrossRef]
- Sloutskin, A.; Kinchington, P.R.; Goldstein, R.S. Productive vs. non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology 2013, 443, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived NeuronsA. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Rajbhandari, L.; Shukla, P.; Jagdish, B.; Lee, H.; Lee, G.; Venkatesan, A. Human stem cell derived sensory neurons are positioned to support varicella zoster virus latency. bioRxiv 2020. [Google Scholar] [CrossRef]
- Christensen, J.; Steain, M.; Slobedman, B.; Abendroth, A. Differentiated neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster virus infection of neuronal cells. J. Virol. 2011, 85, 8436–8442. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.L.; Bowlin, J.L.; Cohrs, R.J.; Gilden, D.; Jones, K.L. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J. Virol. 2014, 88, 5877–5880. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.L.; Zhu, S.; Pearce, C.M.; Viejo-Borbolla, A. Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses 2019, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, L.; Goldstein, R.S.; Kinchington, P.R. Modeling Varicella Zoster Virus Persistence and Reactivation—Closer to Resolving a Perplexing Persistent State. Front. Microbiol. 2019, 10, 1634. [Google Scholar] [CrossRef]
- Aubert, M.; Madden, E.A.; Loprieno, M.; Feelixge, H.S.D.; Stensland, L.; Huang, M.-L.; Greninger, A.L.; Roychoudhury, P.; Niyonzima, N.; Nguyen, T.; et al. In vivo disruption of latent HSV by designer endonuclease therapy. JCI Insight 2016, 1, e88468. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Boyle, N.M.; Stone, D.; Stensland, L.; Huang, M.-L.; Magaret, A.S.; Galetto, R.; Rawlings, D.J.; Scharenberg, A.M.; Jerome, K.R. In vitro Inactivation of Latent HSV by Targeted Mutagenesis Using an HSV-specific Homing Endonuclease. Mol. Ther. Nucleic Acids 2014, 3, e146. [Google Scholar] [CrossRef]
- Aubert, M.; Strongin, D.E.; Roychoudhury, P.; Loprieno, M.A.; Haick, A.K.; Klouser, L.M.; Stensland, L.; Huang, M.-L.; Makhsous, N.; Tait, A.; et al. Gene editing and elimination of latent herpes simplex virus in vivo. Nat. Commun. 2020, 11, 4148. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.G.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.H.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Quake, S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef]
- Dang, C.H.; Aubert, M.; De Silva Feelixge, H.S.; Diem, K.; Loprieno, M.A.; Roychoudhury, P.; Stone, D.; Jerome, K.R. In vivo dynamics of AAV-mediated gene delivery to sensory neurons of the trigeminal ganglia. Sci. Rep. 2017, 7, 927. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80. [Google Scholar] [CrossRef]
- Davison, A.J.; Scott, J.E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986, 67 Pt 9, 1759–1816. [Google Scholar] [CrossRef]
- Davison, A.J.; Scott, J.E. DNA sequence of the major inverted repeat in the varicella-zoster virus genome. J. Gen. Virol. 1985, 66 Pt 2, 207–220. [Google Scholar] [CrossRef]
- Moriuchi, M.; Moriuchi, H.; Straus, S.E.; Cohen, J.I. Varicella-Zoster Virus (VZV) Virion-Associated Transactivator Open Reading Frame 62 Protein Enhances the Infectivity of VZV DNA. Virology 1994, 200, 297–300. [Google Scholar] [CrossRef]
- Kinchington, P.R.; Hougland, J.K.; Arvin, A.M.; Ruyechan, W.T.; Hay, J. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 1992, 66, 359–366. [Google Scholar] [CrossRef]
- Sommer, M.H.; Zagha, E.; Serrano, O.K.; Ku, C.C.; Zerboni, L.; Baiker, A.; Santos, R.; Spengler, M.; Lynch, J.; Grose, C.; et al. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J. Virol. 2001, 75, 8224–8239. [Google Scholar] [CrossRef]
- Sato, B.; Ito, H.; Hinchliffe, S.; Sommer, M.H.; Zerboni, L.; Arvin, A.M. Mutational analysis of open reading frames 62 and 71, encoding the varicella-zoster virus immediate-early transactivating protein, IE62, and effects on replication in vitro and in skin xenografts in the SCID-hu mouse in vivo. J. Virol. 2003, 77, 5607–5620. [Google Scholar] [CrossRef] [PubMed]
- Dremel, S.E.; Deluca, N.A. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. Elife 2019, 8, e51109. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. The varicella-zoster virus genome. Curr. Top. Microbiol. Immunol. 2010, 342, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Felser, J.M.; Kinchington, P.R.; Inchauspe, G.; Straus, S.E.; Ostrove, J.M. Cell lines containing varicella-zoster virus open reading frame 62 and expressing the “IE” 175 protein complement ICP4 mutants of herpes simplex virus type 1. J. Virol. 1988, 62, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Disney, G.H.; Everett, R.D. A herpes simplex virus type 1 recombinant with both copies of the Vmw175 coding sequences replaced by the homologous varicella-zoster virus open reading frame. J. Gen. Virol. 1990, 71 Pt 11, 2681–2689. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, G.P.; Herzog, R.W.; Mount, J.; Arruda, V.R.; Tillson, D.M.; Hathcock, J.; Van Ginkel, F.W.; High, K.A.; Lothrop, C.D., Jr. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 2009, 113, 797–806. [Google Scholar] [CrossRef]
- Harding, T.; Koprivnikar, K.; Tu, G.; Zayek, N.; Lew, S.; Subramanian, A.; Sivakumaran, A.; Frey, D.; Ho, K.; VanRoey, M.; et al. Intravenous administration of an AAV-2 vector for the expression of factor IX in mice and a dog model of hemophilia B. Gene Ther. 2004, 11, 204–213. [Google Scholar] [CrossRef]
- Mount, J.D.; Herzog, R.W.; Tillson, D.M.; Goodman, S.A.; Robinson, N.; McCleland, M.L.; Bellinger, D.; Nichols, T.C.; Arruda, V.R.; Lothrop, C.D., Jr.; et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood 2002, 99, 2670–2676. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; Jenny, M.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Guedon, J.-M.; Zhang, M.; Glorioso, J.; Goins, W.; Kinchington, P. Relief of pain induced by varicella-zoster virus in a rat model of post-herpetic neuralgia using a herpes simplex virus vector expressing enkephalin. Gene Ther. 2014, 21, 694. [Google Scholar] [CrossRef] [PubMed]
- Sloutskin, A.; Goldstein, R.S. Laboratory preparation of Varicella-Zoster Virus: Concentration of virus-containing supernatant, use of a debris fraction and magnetofection for consistent cell-free VZV infections. J. Virol. Methods 2014, 206, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.A.; Stefanovic, T.; Tscharke, D.C. Engineering herpes simplex viruses by infection-transfection methods including recombination site targeting by CRISPR/Cas9 nucleases. J. Virol. Methods 2015, 213, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Choi, V.W.; Asokan, A.; Haberman, R.A.; Samulski, R.J. Production of recombinant adeno-associated viral vectors for in vitro and in vivo use. Curr. Protoc. Mol. Biol. 2007. Chapter 16, Unit 16.25. [Google Scholar] [CrossRef]
- Zolotukhin, S.; Byrne, B.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef]
- Aurnhammer, C.; Haase, M.; Muether, N.; Hausl, M.; Rauschhuber, C.; Huber, I.; Nitschko, H.; Busch, U.; Sing, A.; Ehrhardt, A.; et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 2012, 23, 18–28. [Google Scholar] [CrossRef]
- Tischer, B.K.; Von Einem, J.; Kaufer, B.B.; Osterrieder, N. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Kaufer, B.B.; Sommer, M.H.; Wussow, F.; Arvin, A.M.; Osterrieder, N. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 2007, 81, 13200–13208. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.G.; Smith, N.A.; Tighe, M.; Travis, K.L.; Liu, D.; Upadhyaya, P.K.; Kinchington, P.R.; Chan, G.C.; Moffat, J.F. A Novel Human Skin Tissue Model To Study Varicella-Zoster Virus and Human Cytomegalovirus. J. Virol. 2020, 94, 1082–1102. [Google Scholar] [CrossRef]
- Erazo, A.; Yee, M.B.; Osterrieder, N.; Kinchington, P.R. Varicella-zoster virus open reading frame 66 protein kinase is required for efficient viral growth in primary human corneal stromal fibroblast cells. J. Virol. 2008, 82, 7653–7665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Warner, B.E.; Yee, M.B.; Zhang, M.; Hornung, R.S.; Kaufer, B.B.; Visalli, R.J.; Kramer, P.R.; Goins, W.F.; Kinchington, P.R. Varicella-zoster virus early infection but not complete replication is required for the induction of chronic hypersensitivity in rat models of postherpetic neuralgia. PLoS Pathog. 2021, 17, e1009689. [Google Scholar] [CrossRef]
- Pomp, O.; Brokhman, I.; Ben-Dor, I.; Reubinoff, B.; Goldstein, R.S. Generation of peripheral sensory and sympathetic neurons and neural crest cells from human embryonic stem cells. Stem Cells 2005, 23, 923–930. [Google Scholar] [CrossRef]
- Pomp, O.; Brokhman, I.; Ziegler, L.; Almog, M.; Korngreen, A.; Tavian, M.; Goldstein, R.S. PA6-induced human embryonic stem cell-derived neurospheres: A new source of human peripheral sensory neurons and neural crest cells. Brain Res. 2008, 1230, 50–60. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Turse, S.E.; Jackson, S.A.; Lerner, E.C.; Kinchington, P.R. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J. Virol. 2006, 80, 1710–1723. [Google Scholar] [CrossRef]
- Velusamy, T.; Gowripalan, A.; Tscharke, D.C. CRISPR/Cas9-Based Genome Editing of HSV. Methods Mol. Biol. 2020, 2060, 169–183. [Google Scholar] [CrossRef]
- Jones, J.O.; Sommer, M.; Stamatis, S.; Arvin, A.M. Mutational analysis of the varicella-zoster virus ORF62/63 intergenic region. J. Virol. 2006, 80, 3116–3121. [Google Scholar] [CrossRef][Green Version]
- Hood, C.; Cunningham, A.L.; Slobedman, B.; Arvin, A.M.; Sommer, M.H.; Kinchington, P.R.; Abendroth, A. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 2006, 80, 1025–1031. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Depledge, D.P.; Rajbhandari, L.; Lenac Rovis, T.; Jonjic, S.; Breuer, J.; Venkatesan, A.; Verjans, G.M.G.M.; Sadaoka, T. Varicella-zoster virus VLT-ORF63 fusion transcript induces broad viral gene expression during reactivation from neuronal latency. Nat. Commun. 2020, 11, 6324. [Google Scholar] [CrossRef]
- Ambagala, A.P.N.; Krogmann, T.; Qin, J.; Pesnicak, L.; Cohen, J.I. A varicella-zoster virus mutant impaired for latency in rodents, but not impaired for replication in cell culture. Virology 2010, 399, 194. [Google Scholar] [CrossRef][Green Version]
- Taymans, J.M.; Vandenberghe, L.H.; Van Den Haute, C.; Thiry, I.; Deroose, C.M.; Mortelmans, L.; Wilson, J.M.; Debyser, Z.; Baekelandt, V. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum. Gene Ther. 2007, 18, 195–206. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.; Wilson, J. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef]
- Cearley, C.N.; Wolfe, J.H. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol. Ther. 2006, 13, 528–537. [Google Scholar] [CrossRef]
- Cearley, C.N.; Wolfe, J.H. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J. Neurosci. 2007, 27, 9928–9940. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Deverman, B.E.; Matho, K.S.; Cetin, A.; Woodard, K.; Cepko, C.; Guerin, K.I.; Rego, M.A.; Ersing, I.; Bachle, S.M.; et al. Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 2019, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, A.; Kobinger, G.; Anand, V.; Hildinger, M.; O’Connor, E.; Maguire, A.M.; Wilson, J.M.; Bennett, J. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: The retina as a model. Hum. Mol. Genet. 2001, 10, 3075–3081. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; Stein, C.S.; Heth, J.A.; Martins, I.; Kotin, R.M.; Derksen, T.A.; Zabner, J.; Ghodsi, A.; Chiorini, J.A. Recombinant adeno-associated virus type 2, 4, and 5 vectors: Transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. USA 2000, 97, 3428–3432. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Sloutskin, A.; Markus, A.; Yee, M.B.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus infects human embryonic stem cell-derived neurons and neurospheres but not pluripotent embryonic stem cells or early progenitors. J. Virol. 2012, 86, 3211–3218. [Google Scholar] [CrossRef]
- Lin, H.; Li, G.; Peng, X.; Deng, A.; Ye, L.; Shi, L.; Wang, T.; He, J. The Use of CRISPR/Cas9 as a Tool to Study Human Infectious Viruses. Front. Cell. Infect. Microbiol. 2021, 11, 774. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.; Neuhausser, W.M.; Eggan, P.; Angelova, M.; Kirchner, R.; Eggan, K.C.; Knipe, D.M. Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 2019, 8, e51662. [Google Scholar] [CrossRef] [PubMed]
- Neuhausser, W.; Oh, H.; Eggan, P.; Angelova, M.; Kirchner, R.; Eggan, K.; Knipe, D. Screening Method for CRISPR/Cas9 Inhibition of a Human DNA Virus: Herpes Simplex Virus. Bio-Protoc. 2020, 10, e3748. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Long, K.R.; Loprieno, M.A.; Feelixge, H.S.D.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther. Methods Clin. Dev. 2021, 20, 258. [Google Scholar] [CrossRef]
- Yan, K.; Feng, J.; Liu, X.; Wang, H.; Li, Q.; Li, J.; Xu, T.; Sajid, M.; Ullah, H.; Zhou, L.; et al. Inhibition of Hepatitis B Virus by AAV8-Derived CRISPR/SaCas9 Expressed From Liver-Specific Promoters. Front. Microbiol. 2021, 12, 665184. [Google Scholar] [CrossRef]
- Kunze, C.; Börner, K.; Kienle, E.; Orschmann, T.; Rusha, E.; Schneider, M.; Radivojkov-Blagojevic, M.; Drukker, M.; Desbordes, S.; Grimm, D.; et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia 2018, 66, 413–427. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Chen, Y.C.; Sheng, J.; Trang, P.; Liu, F. Potential Application of the CRISPR/Cas9 System against Herpesvirus Infections. Viruses 2018, 10, 291. [Google Scholar] [CrossRef]
- Yuen, K.S.; Chan, C.P.; Wong, N.H.M.; Ho, C.H.; Ho, T.H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.; Kok, K.H.; et al. CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J. Gen. Virol. 2015, 96, 626–636. [Google Scholar] [CrossRef]
- Adamson, C.S.; Nevels, M.M. Bright and Early: Inhibiting Human Cytomegalovirus by Targeting Major Immediate-Early Gene Expression or Protein Function. Viruses 2020, 12, 110. [Google Scholar] [CrossRef]
- Brackett, K.; Mungale, A.; Lopez-Isidro, M.; Proctor, D.A.; Najarro, G.; Arias, C. CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells. Viruses 2021, 13, 783. [Google Scholar] [CrossRef]
- Akidil, E.; Albanese, M.; Buschle, A.; Ruhle, A.; Pich, D.; Keppler, O.T.; Hammerschmidt, W. Highly efficient CRISPR-Cas9-mediated gene knockout in primary human B cells for functional genetic studies of Epstein-Barr virus infection. PLoS Pathog. 2021, 17, e1009117. [Google Scholar] [CrossRef] [PubMed]
- Braspenning, S.E.; Lebbink, R.J.; Depledge, D.P.; Schapendonk, C.M.E.; Anderson, L.A.; Verjans, G.M.G.M.; Sadaoka, T.; Ouwendijk, W.J.D. Mutagenesis of the varicella-zoster virus genome demonstrates that vlt and vlt-orf63 proteins are dispensable for lytic infection. Viruses 2021, 13, 2289. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.T.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience 2020, 23, 100789. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.T.; Boye, S.L.; Fajardo, D.; Calabro, K.; Peterson, J.J.; Strang, C.E.; Chakraborty, D.; Gloskowski, S.; Haskett, S.; Samuelsson, S.; et al. Somatic Gene Editing of GUCY2D by AAV-CRISPR/Cas9 Alters Retinal Structure and Function in Mouse and Macaque. Hum. Gene Ther. 2019, 30, 571–589. [Google Scholar] [CrossRef]
- Ramlogan-Steel, C.A.; Murali, A.; Andrzejewski, S.; Dhungel, B.; Steel, J.C.; Layton, C.J. Gene therapy and the adeno-associated virus in the treatment of genetic and acquired ophthalmic diseases in humans: Trials, future directions and safety considerations. Clin. Experiment. Ophthalmol. 2019, 47, 521–536. [Google Scholar] [CrossRef]
- Da Costa, B.L.; Levi, S.R.; Eulau, E.; Tsai, Y.-T.; Quinn, P.M.J. Prime Editing for Inherited Retinal Diseases. Front. Genome Ed. 2021, 3, 775330. [Google Scholar] [CrossRef]
- Kedar, S.; Jayagopal, L.N.; Berger, J.R. Neurological and Ophthalmological Manifestations of Varicella Zoster Virus. J. Neuroophthalmol. 2019, 39, 220–231. [Google Scholar] [CrossRef]
- Schoenberger, S.D.; Kim, S.J.; Thorne, J.E.; Mruthyunjaya, P.; Yeh, S.; Bakri, S.J.; Ehlers, J.P. Diagnosis and Treatment of Acute Retinal Necrosis: A Report by the American Academy of Ophthalmology. Ophthalmology 2017, 124, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Nidetz, N.F.; McGee, M.C.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol. Ther. 2020, 207, 107453. [Google Scholar] [CrossRef] [PubMed]
- Goldman, G.S.; King, P.G. Review of the United States universal varicella vaccination program: Herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine 2013, 31, 1680–1694. [Google Scholar] [CrossRef] [PubMed][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Primer Sequence (5′ → 3′) |
|---|---|---|
| ORF62-1 | F | CACCGCTGGTTGAAGTCCCGATACGGA |
| R | AAACTCCGTATCGGGACTTCAACCAGC | |
| ORF62-2 | F | CACCGCCGGCTTTTTACCCGAGATGGA |
| R | AAACTCCATCTCGGGTAAAAAGCCGGC | |
| ORF62-3 | F | CACCGCAGCGCTCTACACCCCAACGCG |
| R | AAACCGCGTTGGGGTGTAGAGCGCTGC | |
| ORF63-1 | F | CACCGATACGCGGGTGCAGAAACCG |
| R | AAACCGGTTTCTGCACCCGCGTATC | |
| ORF63-2 | F | CACCGAAGACGGGTTCATTGAGGCG |
| R | AAACCGCCTCAATGAACCCGTCTTC | |
| ORF63-3 | F | CACCGTTGAATTTCGGGATTCCGACG |
| R | AAACCGTCGGAATCCCGAAATTCAAC | |
| UL3-4 | F | CACCGGTGACGAGCGCGATCCGGC |
| R | AAACGCCGGATCGCGCTCGTCACC |
| Gene | Direction | Primer Sequence (5′ → 3′) |
|---|---|---|
| SaCas9 | L | AGAAATACGTGGCCGAACTG |
| R | TCACGTAGTCGCTGGTCTTG | |
| ORF49 (VZV genome) | F | CGGTCGAGGAGGAATCTGTG |
| R | CCGTTGCACGTAACAAGCTC |
| Gene | Direction | |
|---|---|---|
| 62 gRNA region primers (5′ → 3′) a | F | GTCATGGTGGGACGGGAACATGAGATCGTTTCAATTCCCagtGTcagtGGcCTgCAgCCtGAACCCAGAAGGATGACTACGATAAGTAGG |
| R | TTGTGTTAGCTCTTCGCCAACATCTTCCGTTCTGGGTTCaGGcTGcAGgCCactgACactGGGAATTGAAAGGGTAATGCCAGTGTTAC | |
| Wobble base pair changes b | VZV DR | TCC GTA TCG GGA CTT CAA CCA G → S68 V69 S70 G71 L72 Q73 P74 |
| VZV DR-62gmut | agt GTc agt GGc CTg CAg CCt G → S68 V69 S70 G71 L72 Q73 P74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.W.; Yee, M.B.; Goldstein, R.S.; Kinchington, P.R. Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71. Viruses 2022, 14, 378. https://doi.org/10.3390/v14020378
Wu BW, Yee MB, Goldstein RS, Kinchington PR. Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71. Viruses. 2022; 14(2):378. https://doi.org/10.3390/v14020378
Chicago/Turabian StyleWu, Betty W., Michael B. Yee, Ronald S. Goldstein, and Paul R. Kinchington. 2022. "Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71" Viruses 14, no. 2: 378. https://doi.org/10.3390/v14020378
APA StyleWu, B. W., Yee, M. B., Goldstein, R. S., & Kinchington, P. R. (2022). Antiviral Targeting of Varicella Zoster Virus Replication and Neuronal Reactivation Using CRISPR/Cas9 Cleavage of the Duplicated Open Reading Frames 62/71. Viruses, 14(2), 378. https://doi.org/10.3390/v14020378