
PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Creation
2.2. Feature Generation
2.3. Training and Testing Using XGBoost
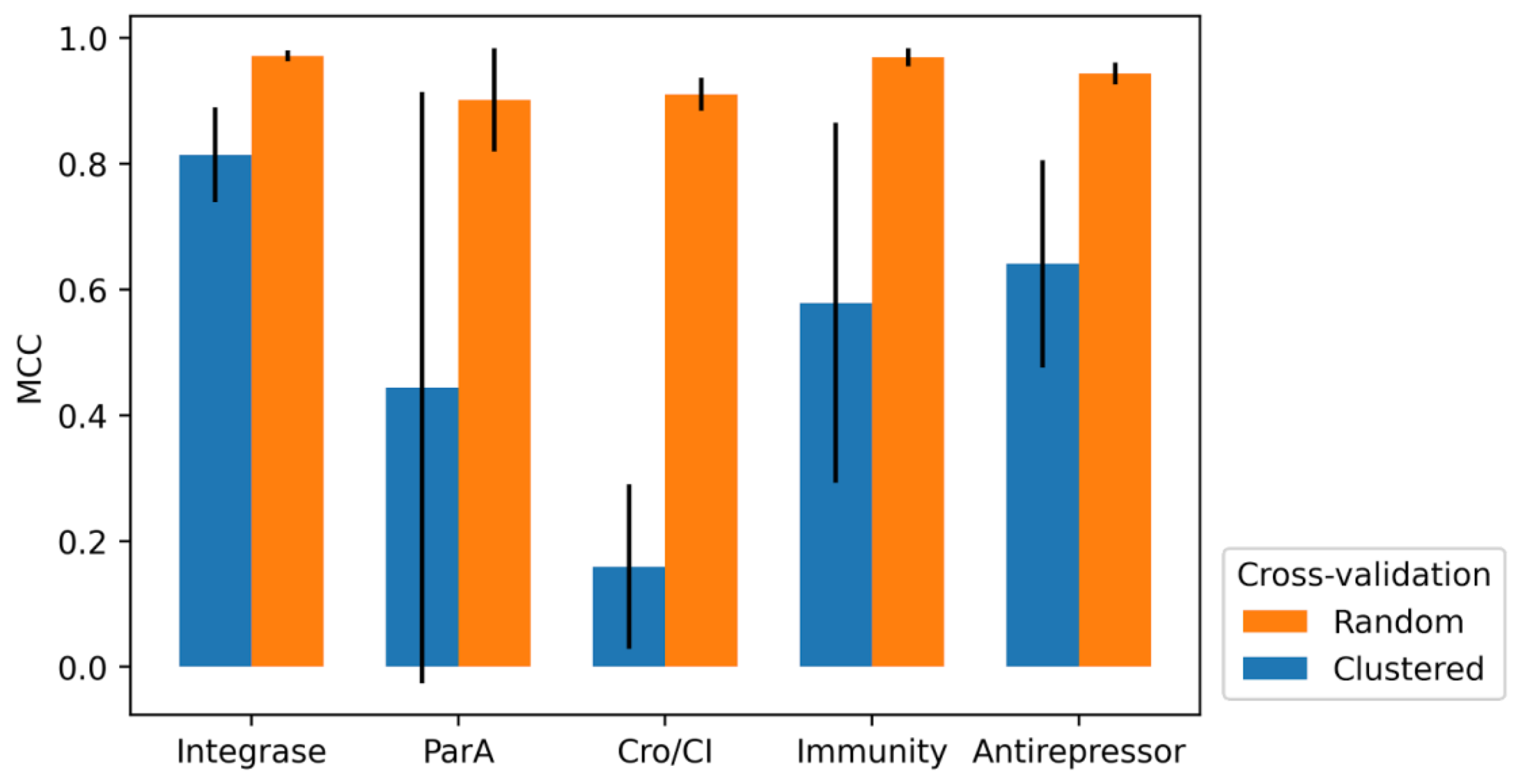
2.4. 10-Fold Cross-Validation
2.5. Screening for Virulence and Undesirable Genes
3. Results and Discussions
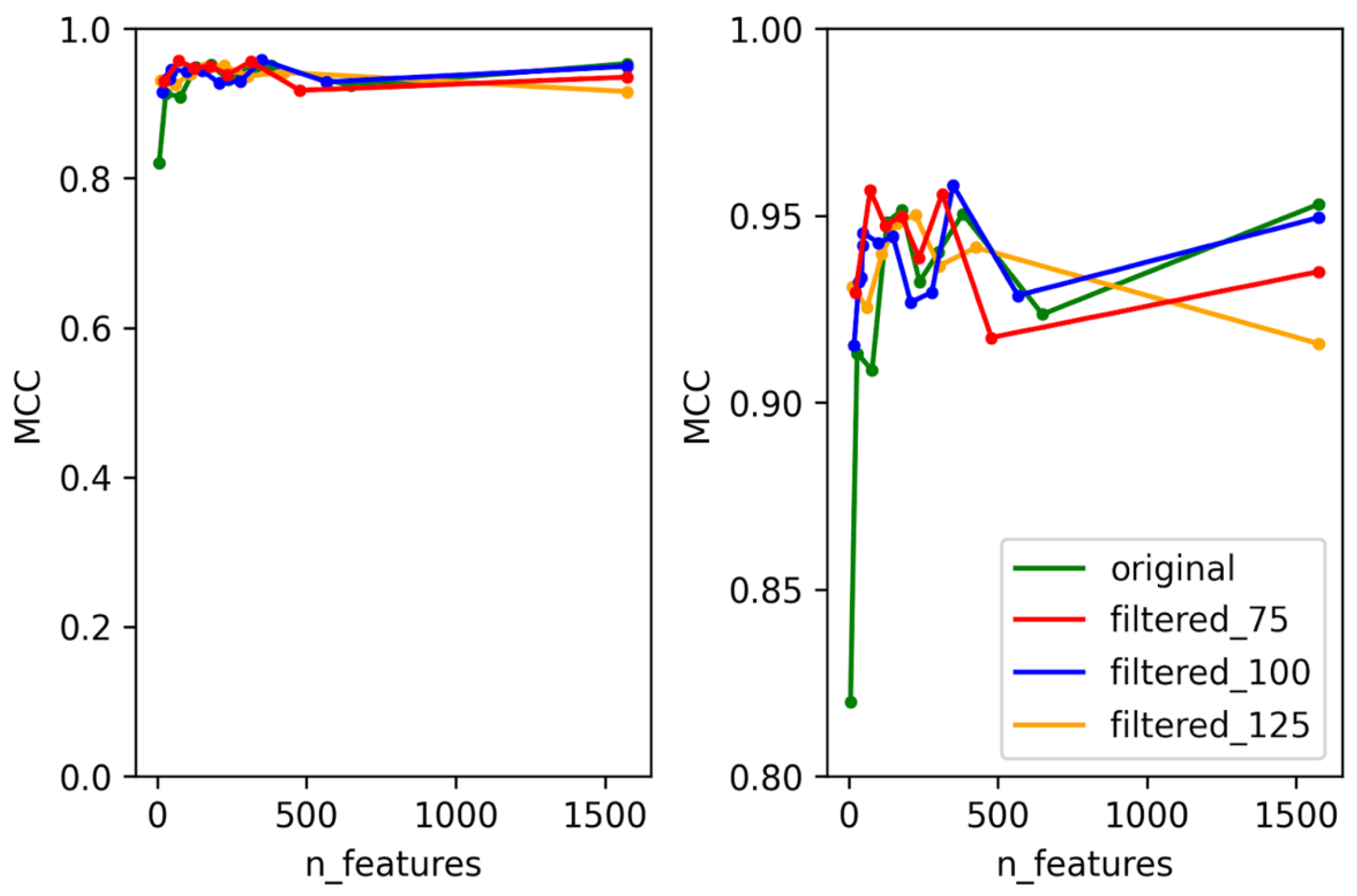
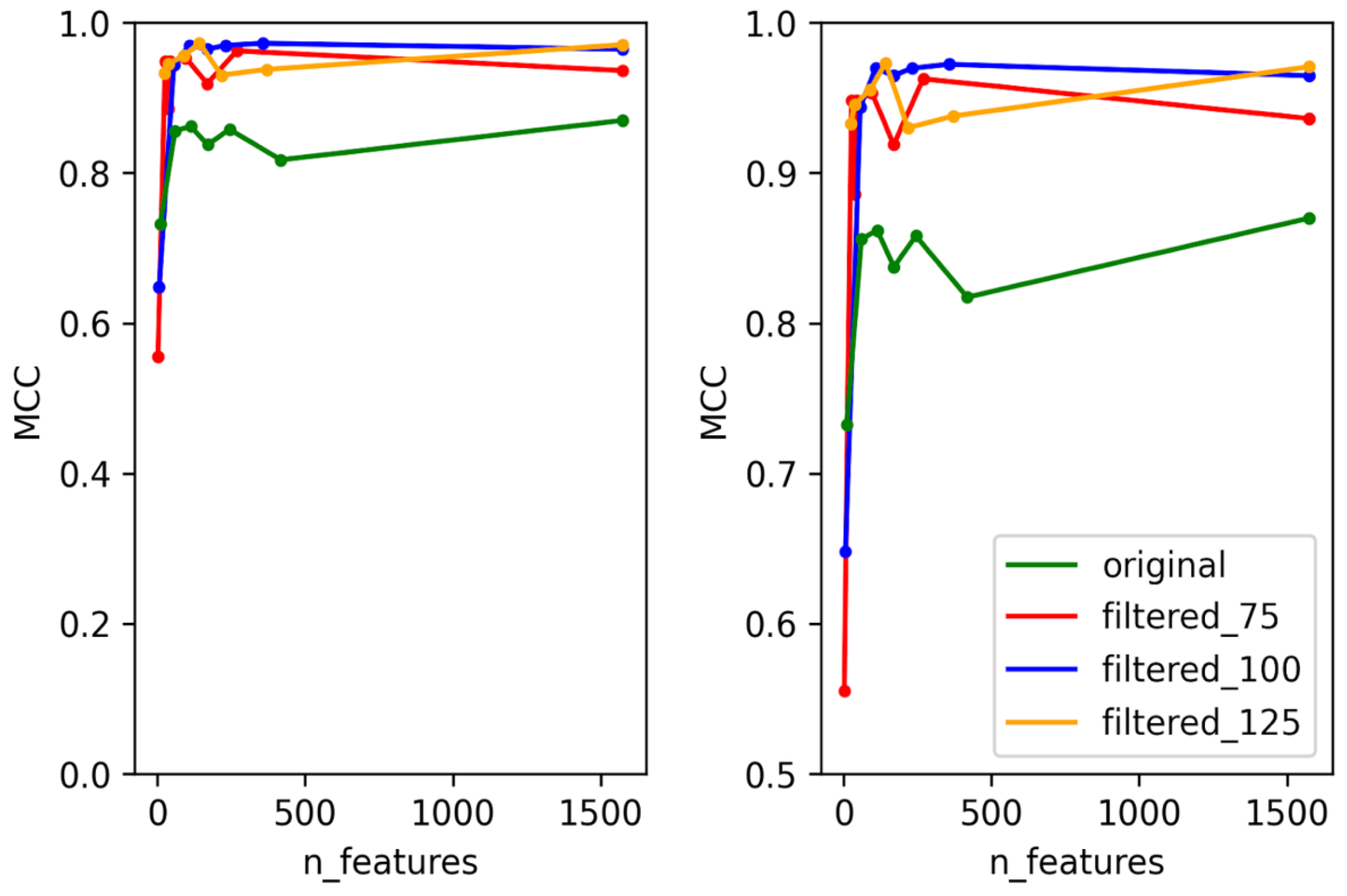
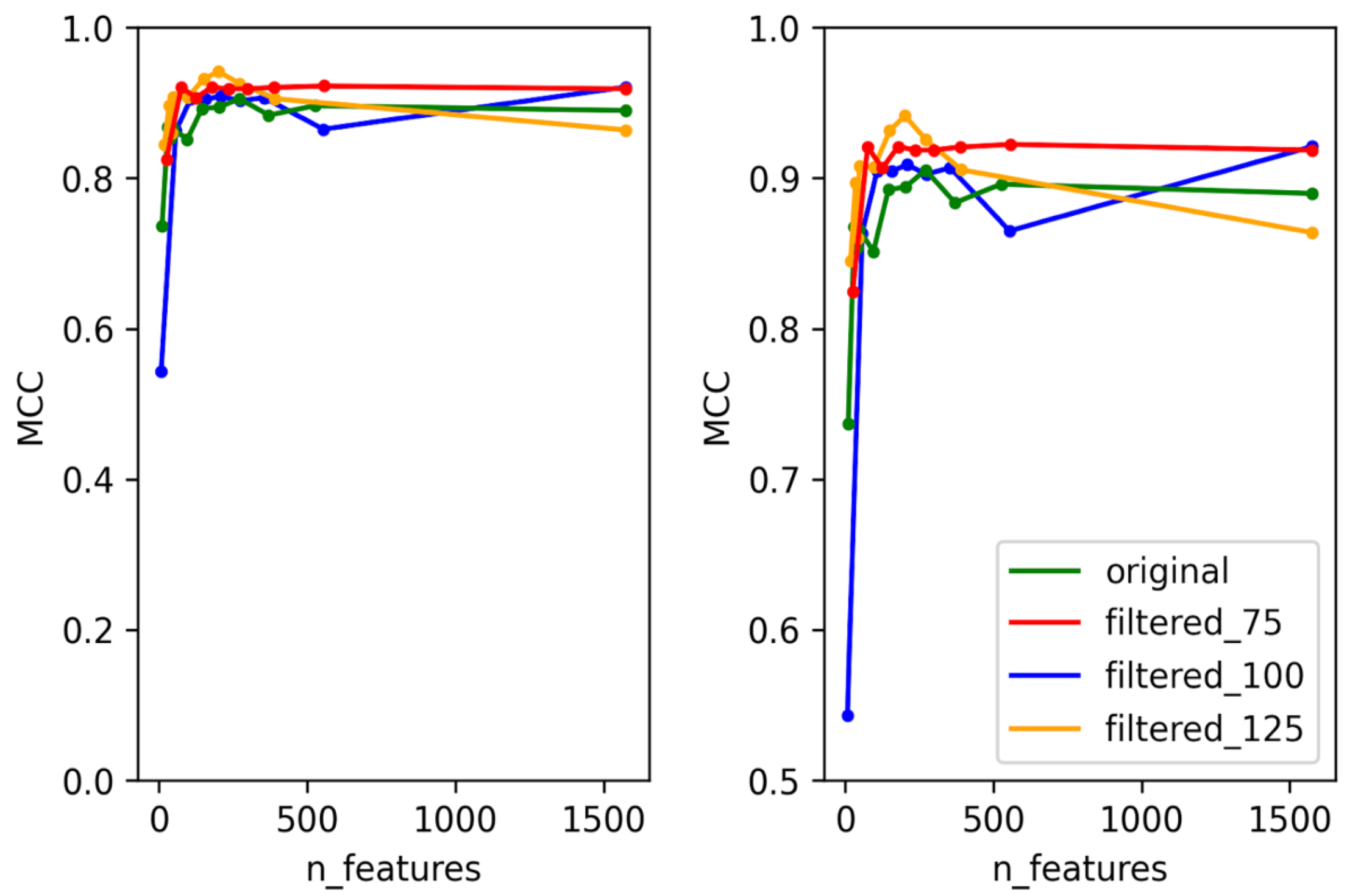
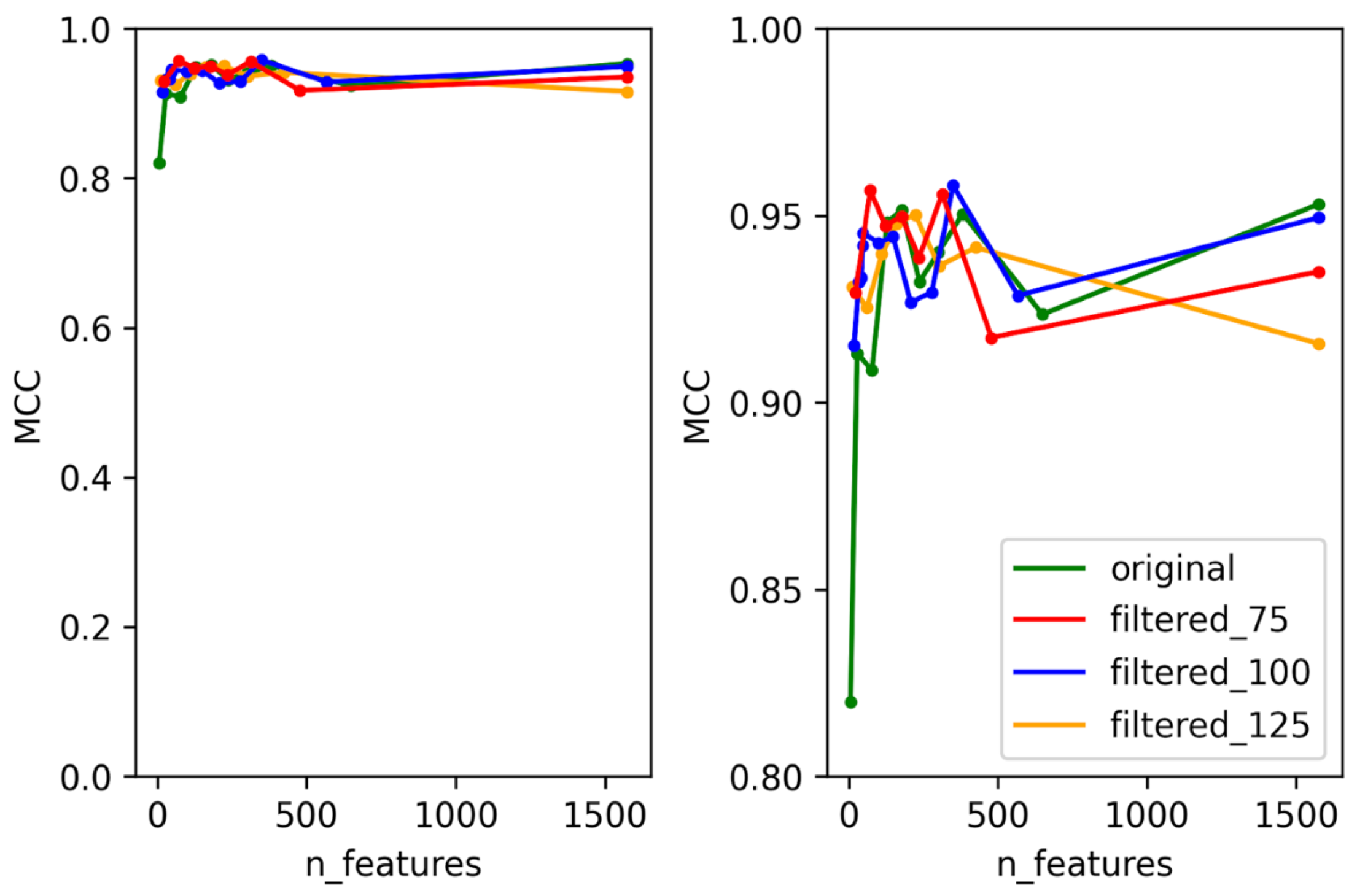
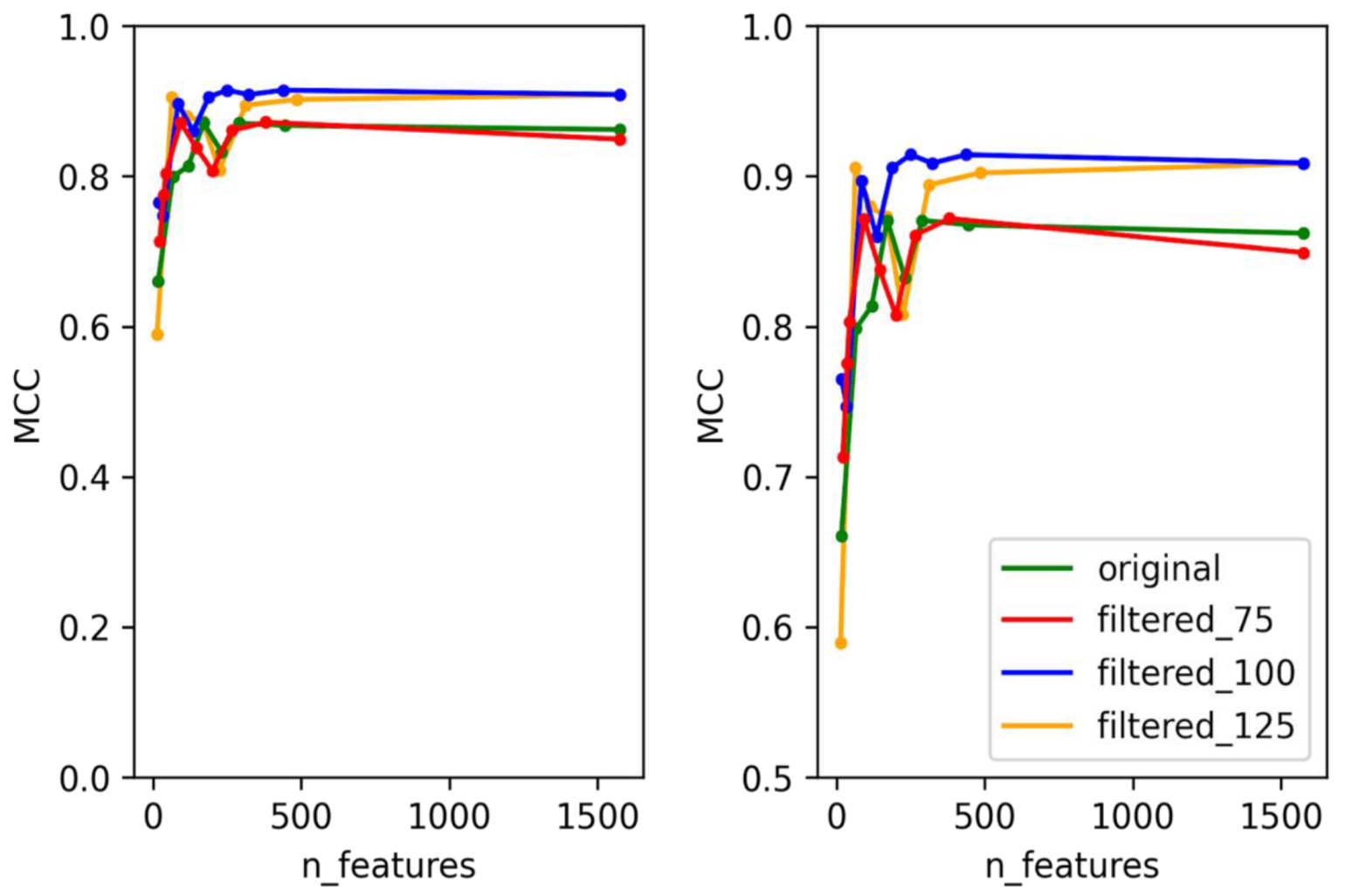
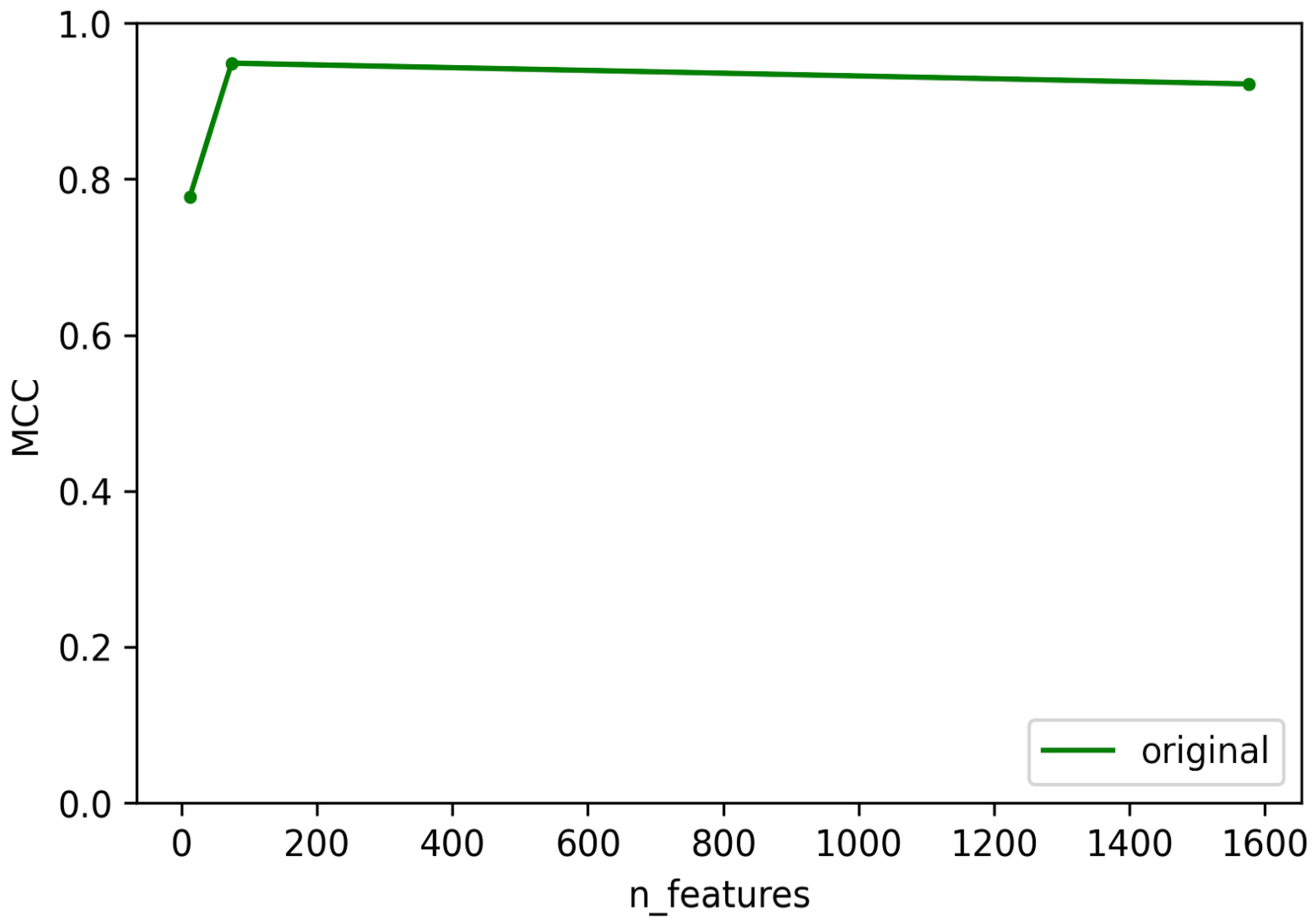
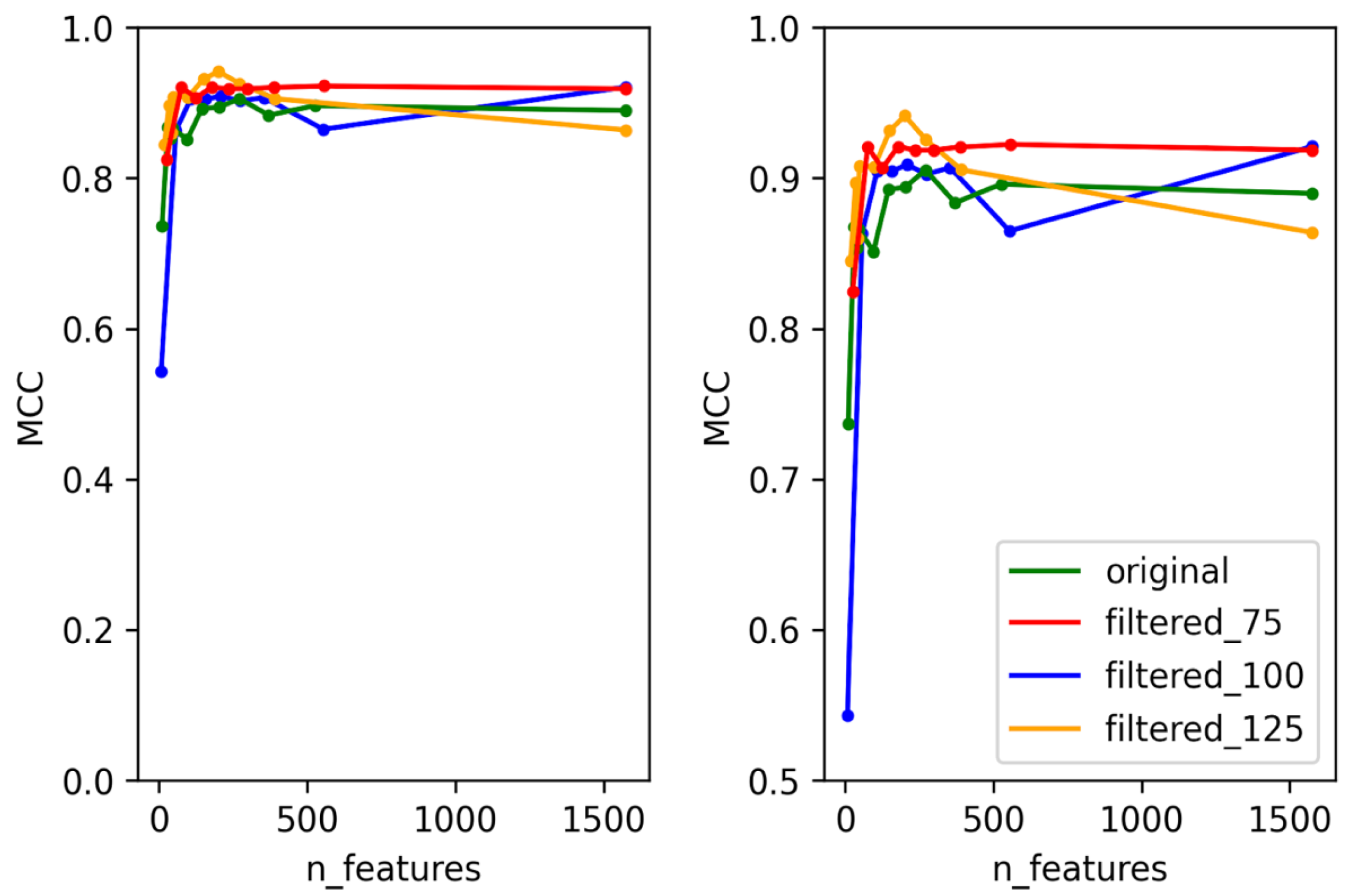
3.1. Selection of Best Dataset and Best Features
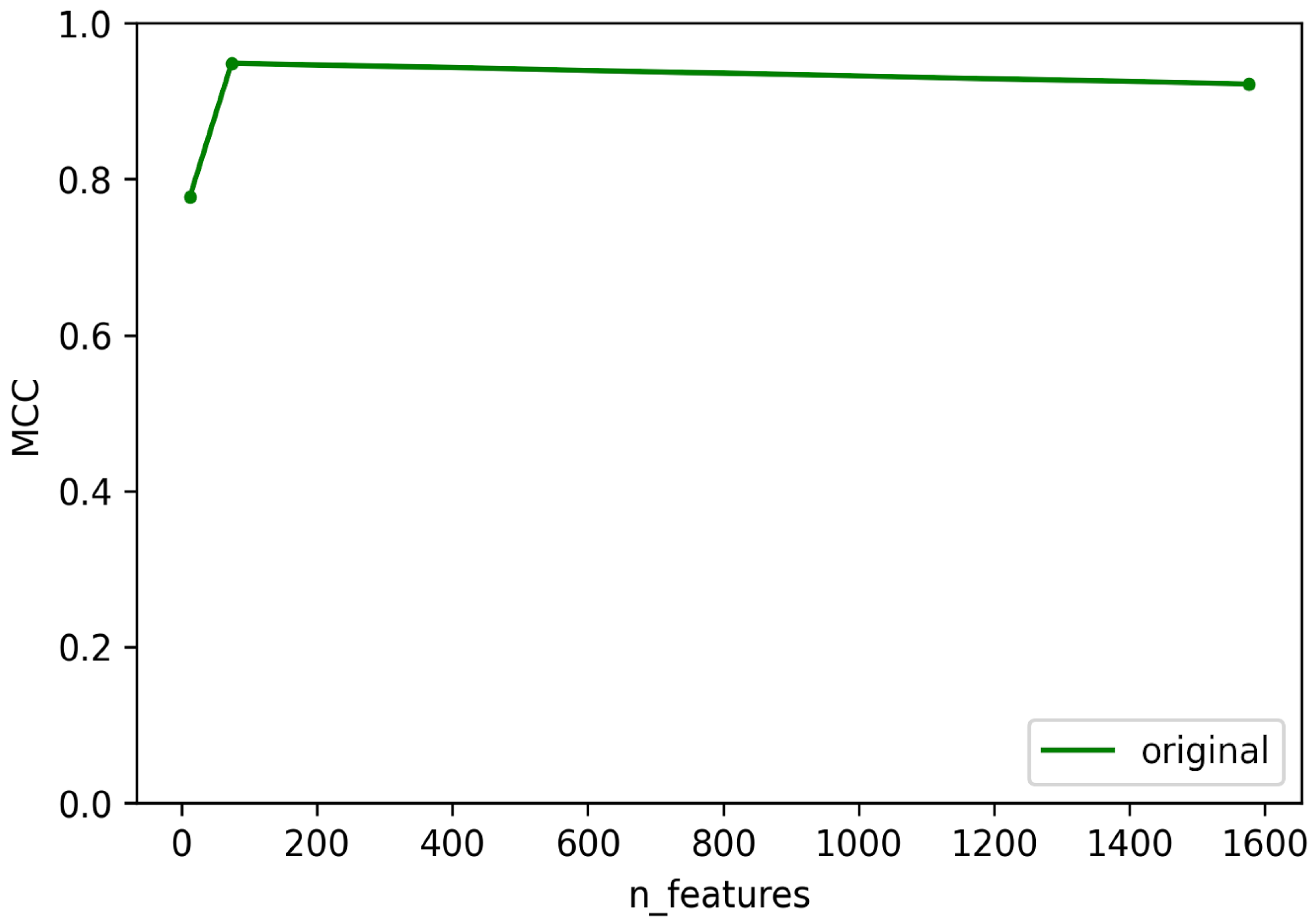
3.2. Optimization and Validation of Final Predictor
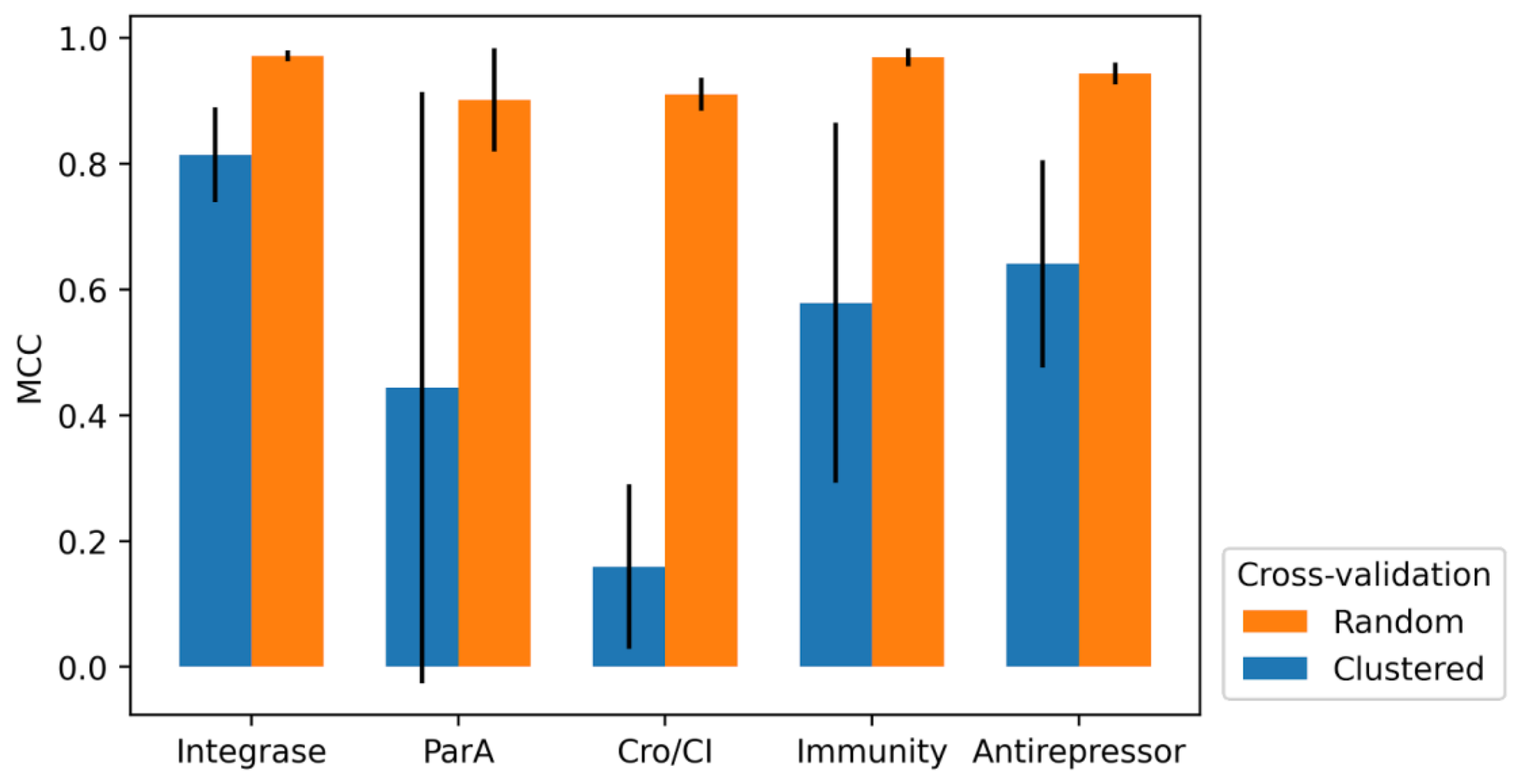
3.3. 10-Fold Cross-Validation
3.4. Feature Importance
3.5. Prediction of Phage Lifestyle
3.6. Caveat in Phage Protein Predictors
3.7. AMR and Virulence Genes in Lytic Phages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundin, G.W.; Bender, C.L. Dissemination of the StrA-StrB Streptomycin-Resistance Genes among Commensal and Pathogenic Bacteria from Humans, Animals, and Plants. Mol. Ecol. 1996, 5, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling Antimicrobial Resistance. Curr. Med. Chem. 2017, 24, 4303–4314. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Kolenda, C.; Batailler, C.; Gustave, C.-A.; Lustig, S.; Malatray, M.; Fevre, C.; Josse, J.; Petitjean, C.; Chidiac, C.; et al. Phage Therapy as Adjuvant to Conservative Surgery and Antibiotics to Salvage Patients with Relapsing, S. Aureus Prosthetic Knee Infection. Front. Med. 2020, 7, 570572. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and Tolerability of a Cocktail of Bacteriophages to Treat Burn Wounds Infected by Pseudomonas Aeruginosa (PhagoBurn): A Randomised, Controlled, Double-Blind Phase 1/2 Trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Aslam, S.; Lampley, E.; Wooten, D.; Karris, M.; Benson, C.; Strathdee, S.; Schooley, R.T. Lessons Learned From the First 10 Consecutive Cases of Intravenous Bacteriophage Therapy to Treat Multidrug-Resistant Bacterial Infections at a Single Center in the United States. Open Forum Infect. Dis. 2020, 7, ofaa389. [Google Scholar] [CrossRef]
- Leitner, L.; Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Chkonia, I.; Rigvava, S.; Chkhotua, A.; Changashvili, G.; McCallin, S.; Schneider, M.P.; et al. Intravesical Bacteriophages for Treating Urinary Tract Infections in Patients Undergoing Transurethral Resection of the Prostate: A Randomised, Placebo-Controlled, Double-Blind Clinical Trial. Lancet Infect. Dis. 2021, 21, 427–436. [Google Scholar] [CrossRef]
- Onsea, J.; Soentjens, P.; Djebara, S.; Merabishvili, M.; Depypere, M.; Spriet, I.; De Munter, P.; Debaveye, Y.; Nijs, S.; Vanderschot, P.; et al. Bacteriophage Application for Difficult-to-Treat Musculoskeletal Infections: Development of a Standardized Multidisciplinary Treatment Protocol. Viruses 2019, 11, 891. [Google Scholar] [CrossRef] [Green Version]
- Doub, J.B. Bacteriophage Therapy for Clinical Biofilm Infections: Parameters That Influence Treatment Protocols and Current Treatment Approaches. Antibiotics 2020, 9, 799. [Google Scholar] [CrossRef]
- Philipson, C.W.; Voegtly, L.J.; Lueder, M.R.; Long, K.A.; Rice, G.K.; Frey, K.G.; Biswas, B.; Cer, R.Z.; Hamilton, T.; Bishop-Lilly, K.A. Characterizing Phage Genomes for Therapeutic Applications. Viruses 2018, 10, 188. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Hendrix, H.; Skurnik, M.; Lavigne, R. Phage-Based Target Discovery and Its Exploitation towards Novel Antibacterial Molecules. Curr. Opin. Biotechnol. 2021, 68, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate Representation of Evolutionary and Functional Relationships between Phage Genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Doub, J.B. Risk of Bacteriophage Therapeutics to Transfer Genetic Material and Contain Contaminants Beyond Endotoxins with Clinically Relevant Mitigation Strategies. Infect. Drug Resist. 2021, 14, 5629–5637. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.J.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in Nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Mäntynen, S.; Laanto, E.; Oksanen, H.M.; Poranen, M.M.; Díaz-Muñoz, S.L. Black Box of Phage–Bacterium Interactions: Exploring Alternative Phage Infection Strategies. Open Biol. 2021, 11, 210188. [Google Scholar] [CrossRef] [PubMed]
- Colavecchio, A.; Cadieux, B.; Lo, A.; Goodridge, L.D. Bacteriophages Contribute to the Spread of Antibiotic Resistance Genes among Foodborne Pathogens of the Enterobacteriaceae Family—A Review. Front. Microbiol. 2017, 8, 1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically Aware Phage Therapy: Pharmacodynamic and Pharmacokinetic Obstacles to Phage Antibacterial Action in Animal and Human Bodies. Microbiol. Mol. Biol. Rev. 2019, 83, e00012-19. [Google Scholar] [CrossRef]
- Moon, K.; Jeon, J.H.; Kang, I.; Park, K.S.; Lee, K.; Cha, C.-J.; Lee, S.H.; Cho, J.-C. Freshwater Viral Metagenome Reveals Novel and Functional Phage-Borne Antibiotic Resistance Genes. Microbiome 2020, 8, 75. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.Y.; Lin, J.T.; Ricker, N.; Anany, H. The Age of Phage: Friend or Foe in the New Dawn of Therapeutic and Biocontrol Applications? Pharmaceuticals 2021, 14, 199. [Google Scholar] [CrossRef]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a Computational Approach to Classifying the Lifestyle of Phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting Bacteriophage Lifestyle from Conserved Protein Domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in Homology Search: HMMER3 and Convergent Evolution of Coiled-Coil Regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charoenkwan, P.; Kanthawong, S.; Schaduangrat, N.; Yana, J.; Shoombuatong, W. PVPred-SCM: Improved Prediction and Analysis of Phage Virion Proteins Using a Scoring Card Method. Cells 2020, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a Fast and Accurate Tool and Web Server to Classify Phage Structural Proteins. PLOS Comput. Biol. 2020, 16, e1007845. [Google Scholar] [CrossRef]
- Sirén, K.; Millard, A.; Petersen, B.; Gilbert, M.T.P.; Clokie, M.R.J.; Sicheritz-Pontén, T. Rapid Discovery of Novel Prophages Using Biological Feature Engineering and Machine Learning. NAR Genom. Bioinforma. 2021, 3, lqaa109. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Kaminski, J.; Gibson, M.K.; Franzosa, E.A.; Segata, N.; Dantas, G.; Huttenhower, C. High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLOS Comput. Biol. 2015, 11, e1004557. [Google Scholar] [CrossRef] [Green Version]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A Database for Classification of Antimicrobial Drug, Biocide and Metal Resistance Determinants in Metagenomic Sequence Data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- National Database of Antibiotic Resistant Organisms (NDARO)—Pathogen Detection—NCBI. Available online: https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/ (accessed on 17 November 2021).
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis--10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. ABRicate; Melbourne, Australia, 2021; Available online: https://github.com/tseemann/abricate (accessed on 20 January 2022).
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.; Swart, P.; S Chult, D. Exploring Network Structure, Dynamics, and Function Using Networkx. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.-H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In Silico Serotyping of E. Coli from Short Read Data Identifies Limited Novel O-Loci but Extensive Diversity of O:H Serotype Combinations within and between Pathogenic Lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The Advantages of the Matthews Correlation Coefficient (MCC) over F1 Score and Accuracy in Binary Classification Evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ofer, D.; Linial, M. ProFET: Feature Engineering Captures High-Level Protein Functions. Bioinformatics 2015, 31, 3429–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z. Propy: A Tool to Generate Various Modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanni, L.; Lumini, A.; Brahnam, S. An Empirical Study of Different Approaches for Protein Classification. Sci. World J. 2014, 2014, e236717. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.A.; Reinders, M.J.; Roubos, J.A.; Ridder, D. de SPiCE: A Web-Based Tool for Sequence-Based Protein Classification and Exploration. BMC Bioinform. 2014, 15, 93. [Google Scholar] [CrossRef] [Green Version]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage Evolution Differs by Host, Lifestyle and Genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef] [Green Version]
- Shitrit, D.; Hackl, T.; Laurenceau, R.; Raho, N.; Carlson, M.C.G.; Sabehi, G.; Schwartz, D.A.; Chisholm, S.W.; Lindell, D. Genetic Engineering of Marine Cyanophages Reveals Integration but Not Lysogeny in T7-like Cyanophages. ISME J. 2021, 16, 488–499. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Keywords Used for Positive Labels | Number of Positive Labels | Number of Negative Labels |
|---|---|---|---|
| Integrase | Integrase, site-specific recombinase, tyrosine recombinase, serine recombinase, int, tyr recombinase, ser recombinase | Training: 2224 Validation: 485 | Training: 156,132 Validation: 38,885 |
| Cro/CI | Cro, CI, C1, CL | Training: 657 Validation: 132 | Training: 156,170 Validation: 39,238 |
| Immunity repressor | Immunity related repressor, immunity repressor, ImmR | Training: 754 Validation: 305 | Training: 157,602 Validation: 39,065 |
| ParA | DNA partitioning protein A, Chromosome partitioning protein A, ParA, partitioning protein A | Training: 78 Validation: 9 | Training: 158,663 Validation: 39,435 |
| Antirepressor | Antirepressor, anti-repressor, antirepressor Rha, Rha | Training: 945 Validation: 185 | Training: 157,864 Validation: 39,275 |
| Predictors | Threshold | Validation MCC | Validation F1 | Validation Accuracy |
|---|---|---|---|---|
| Integrase | 0.252 | 0.92 | 0.92 | 0.99 |
| Cro/CI | 0.363 | 0.71 | 0.71 | 0.99 |
| Immunity repressor | 0.472 | 0.87 | 0.86 | 0.99 |
| ParA | 0.03 | 0.76 | 0.75 | 0.99 |
| Antirepressor | 0.389 | 0.92 | 0.92 | 0.99 |
| Lytic | Temperate | |
|---|---|---|
| Lytic | 177 | 7 |
| Temperate | 9 | 231 |
| Phage Genome | Protein Name | Protein ID | Predicted Marker | Remark |
|---|---|---|---|---|
| Salmonella phage SPN19 (NC_019417) | hypothetical protein | YP_006990261.1 | Integrase | Blastp result shows high similarity (99.1%) to viral integrase family 4 [Salmonella Phage 37;YP_009221453.1] |
| Salmonella phage FSL SP-088 (NC_021780) | hypothetical protein | YP_008239914.1 | Integrase | Blastp result shows high similarity (97.3%) to viral integrase family 4 [Salmonella Phage 37;YP_009221453.1] |
| Lactococcus phage 4268 (NC_004746) | DUF739 family protein | NP_839893.1 | Cro/CI | DUF739/pfam05339 contains putative Cro/CI repressors |
| helix-turn-helix transcriptional regulator | NP_839899.1 | Cro/CI | HTH-transcriptional regulator/repressor/HTH -containing proteins are common ambiguous annotations of Cro/CI | |
| putative antirepressor | NP_839894 | Antirepressor | Lytic phage with lysogeny marker | |
| Lactobacillus phage Lc-Nu (NC_007501) | CI-like repressor | YP_358780.1 | Cro/CI | Lytic phage with lysogeny marker |
| Cro-like repressor | YP_358781.1 | Cro/CI | Lytic phage with lysogeny marker | |
| Mycobacterium phage LRRHood (GQ303262) | immunity repressor | ACU41572.1 | Immunity repressor | Lytic phage with lysogeny marker |
| Mycobacterium phage Alice (JF704092) | hypothetical protein | AEJ94305 | Immunity repressor | Blastp result shows high similarity(98.82%) to immunity repressor [Mycobacterium phage Phox; ATN91327.1] |
| Phage Genome | Gene | Product | Resistance/Virulence | Database Source |
|---|---|---|---|---|
| Stx2 converting phage vB_EcoP_24B (NC_027984) | CatA1 | type A-1 chloramphenicol O-acetyltransferase | Chloramphenicol resistance | NCBI, CARD, ARGANNOT, resfinder, megares |
| iss2 | Increase serum survival protein | Virulence | ecoli_vf | |
| Enterobacteria phage lambda (NC_001416) | iss2 | Increase serum survival protein | Virulence | ecoli_vf |
| Phage cdtI (NC_009514) | nleH1 | Type III secretion system effector NleH1 | Virulence | ecoli_vf |
| cif | Type III secretion system effector Cif cyclomodulin | Virulence | ecoli_vf | |
| cdtA | Cytolethal distending toxin subunit A | Virulence | ecoli_vf | |
| cdtB | Cytolethal distending toxin subunit B | Virulence | ecoli_vf | |
| cdtC | Cytolethal distending toxin subunit C | Virulence | ecoli_vf | |
| Enterobacteria phage YYZ-2008 (NC_011356) | stx1A | Shiga toxin 1 subunit A | Virulence | ecoli_vf |
| stx1B | Shiga-like toxin 1 subunit B | Virulence | ecoli_vf | |
| nleG6-3 | NleG Type 3 Effectors | Virulence | ecoli_vf | |
| nleG5-1 | NleG Type 1 Effectors | Virulence | ecoli_vf | |
| Escherichia phage TL-2011c (NC_019442) | stx2A | Shiga toxin 2 subunit A | Virulence | ecoli_vf |
| stx2B | Shiga-like toxin II subunit B | Virulence | ecoli_vf | |
| iss2 | Increase serum survival protein | Virulence | ecoli_vf | |
| Enterobacteria phage HK629 (NC_019711) | iss2 | Increase serum survival protein | Virulence | ecoli_vf |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yukgehnaish, K.; Rajandas, H.; Parimannan, S.; Manickam, R.; Marimuthu, K.; Petersen, B.; Clokie, M.R.J.; Millard, A.; Sicheritz-Pontén, T. PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach. Viruses 2022, 14, 342. https://doi.org/10.3390/v14020342
Yukgehnaish K, Rajandas H, Parimannan S, Manickam R, Marimuthu K, Petersen B, Clokie MRJ, Millard A, Sicheritz-Pontén T. PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach. Viruses. 2022; 14(2):342. https://doi.org/10.3390/v14020342
Chicago/Turabian StyleYukgehnaish, Kumarasan, Heera Rajandas, Sivachandran Parimannan, Ravichandran Manickam, Kasi Marimuthu, Bent Petersen, Martha R. J. Clokie, Andrew Millard, and Thomas Sicheritz-Pontén. 2022. "PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach" Viruses 14, no. 2: 342. https://doi.org/10.3390/v14020342
APA StyleYukgehnaish, K., Rajandas, H., Parimannan, S., Manickam, R., Marimuthu, K., Petersen, B., Clokie, M. R. J., Millard, A., & Sicheritz-Pontén, T. (2022). PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach. Viruses, 14(2), 342. https://doi.org/10.3390/v14020342