
DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Plasmids, Reagents, and Viruses
2.2. CRISPR-Cas9 Generation of Knockout Cell Lines
2.3. pLDT and pCW57 Cell Line Generation
2.4. Luciferase Reporter Assay
2.5. Retroviral Transduction and Stable Knockdown Cell Lines
2.6. ELISAs and RT-qPCR
2.7. Immunoprecipitations
2.8. Immunoblotting
2.9. Virus Growth Assays
2.10. Cell Sub-Fractionation
2.11. Immunofluorescence
2.12. Statistics
3. Results
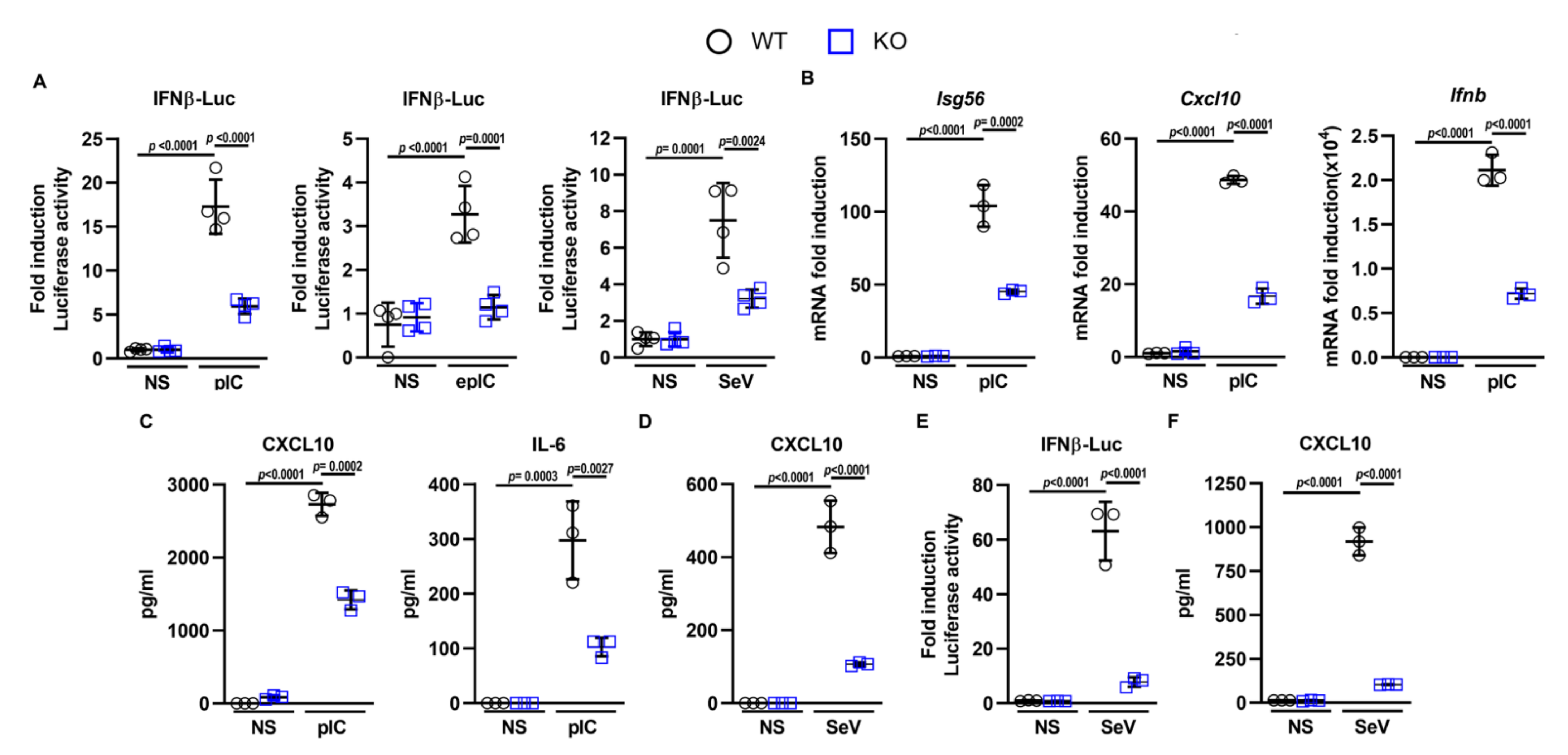
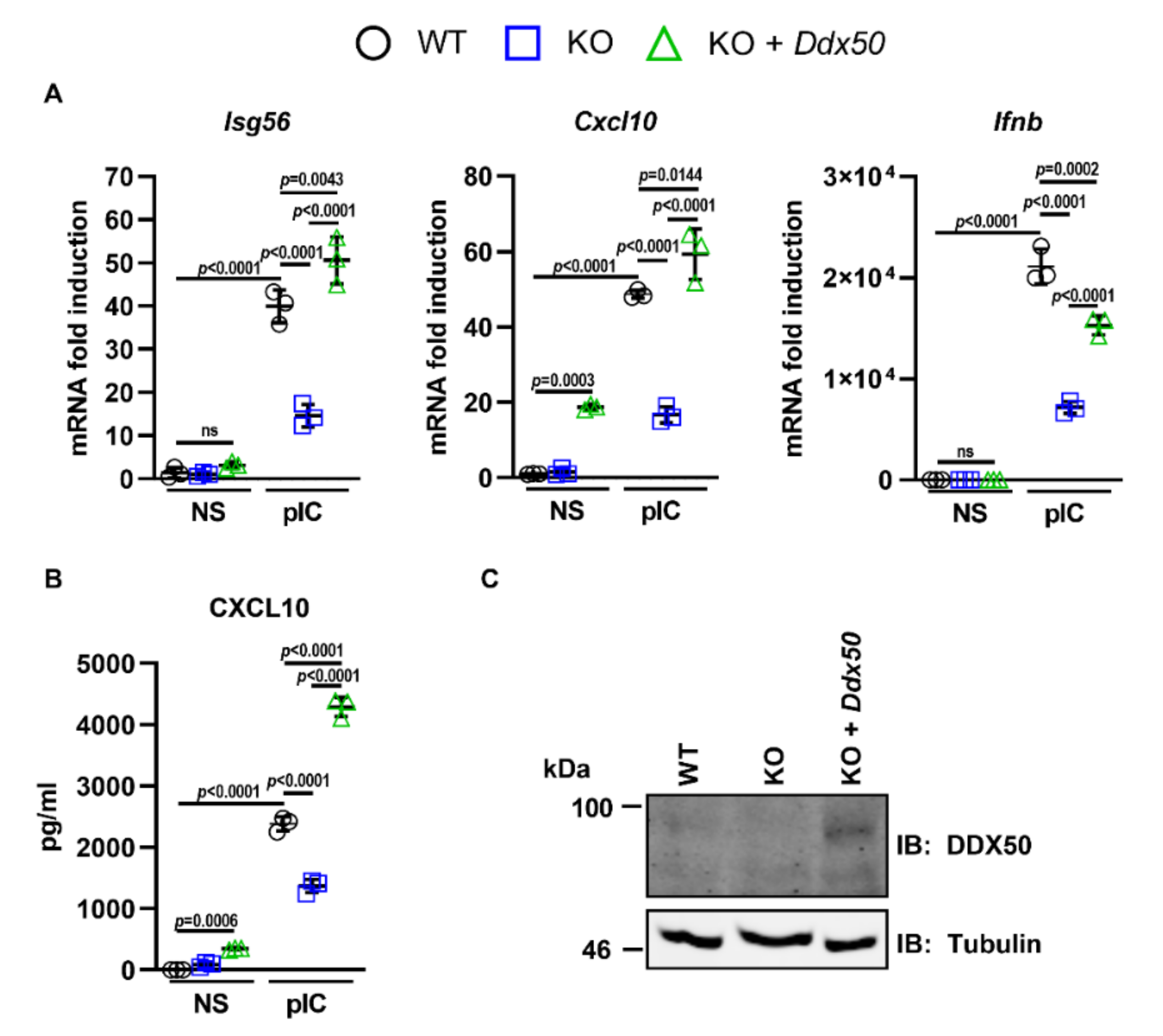
3.1. DDX50 Is a Novel Factor Required for the Innate Immune Response to Nucleic Acid
3.2. Loss of Ddx50 Does Not Alter IL-1α or TNFα-Mediated NF-κB Activation
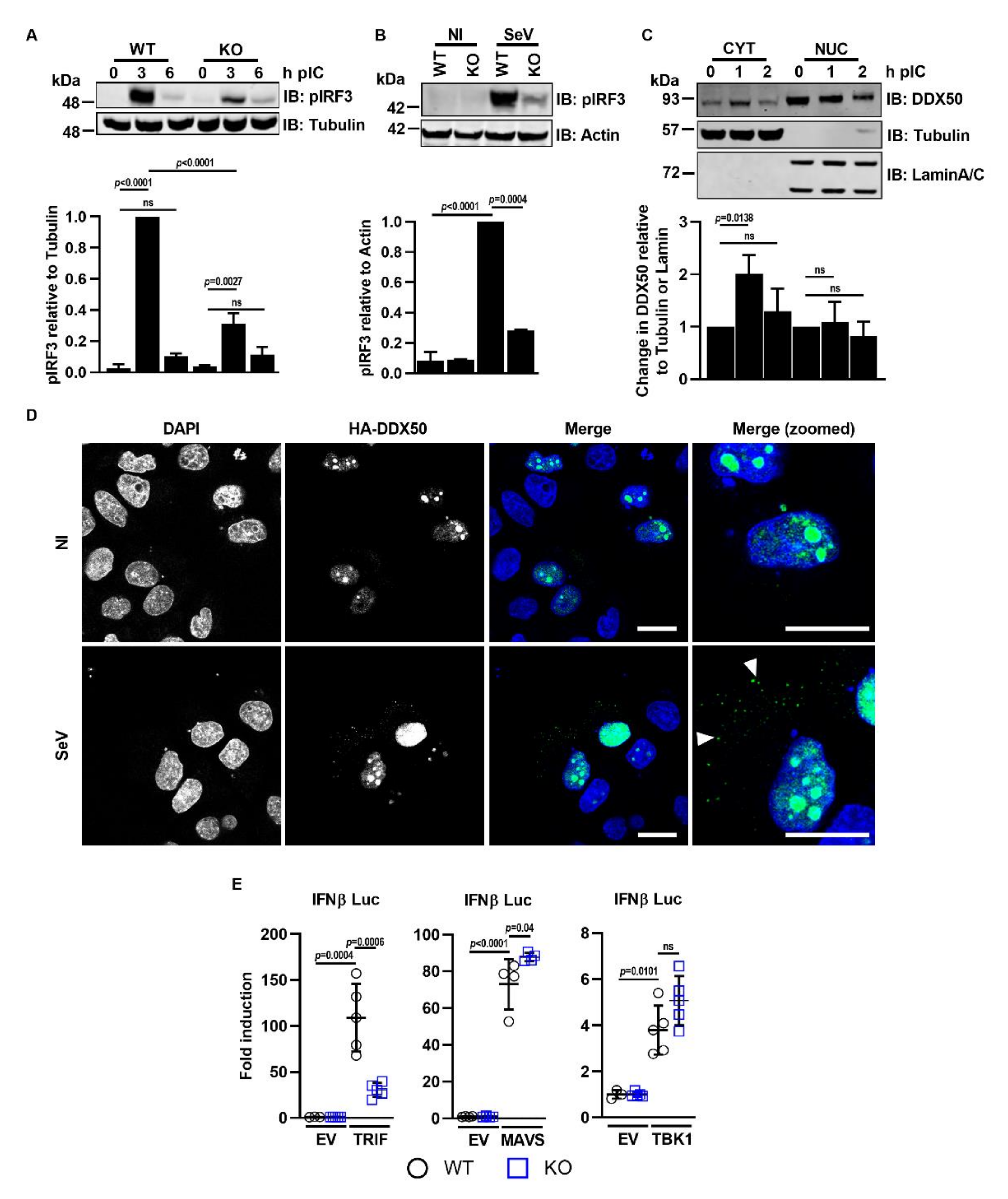
3.3. DDX50 Accumulates in the Cytoplasm to Activate Signalling Upstream of MAVS
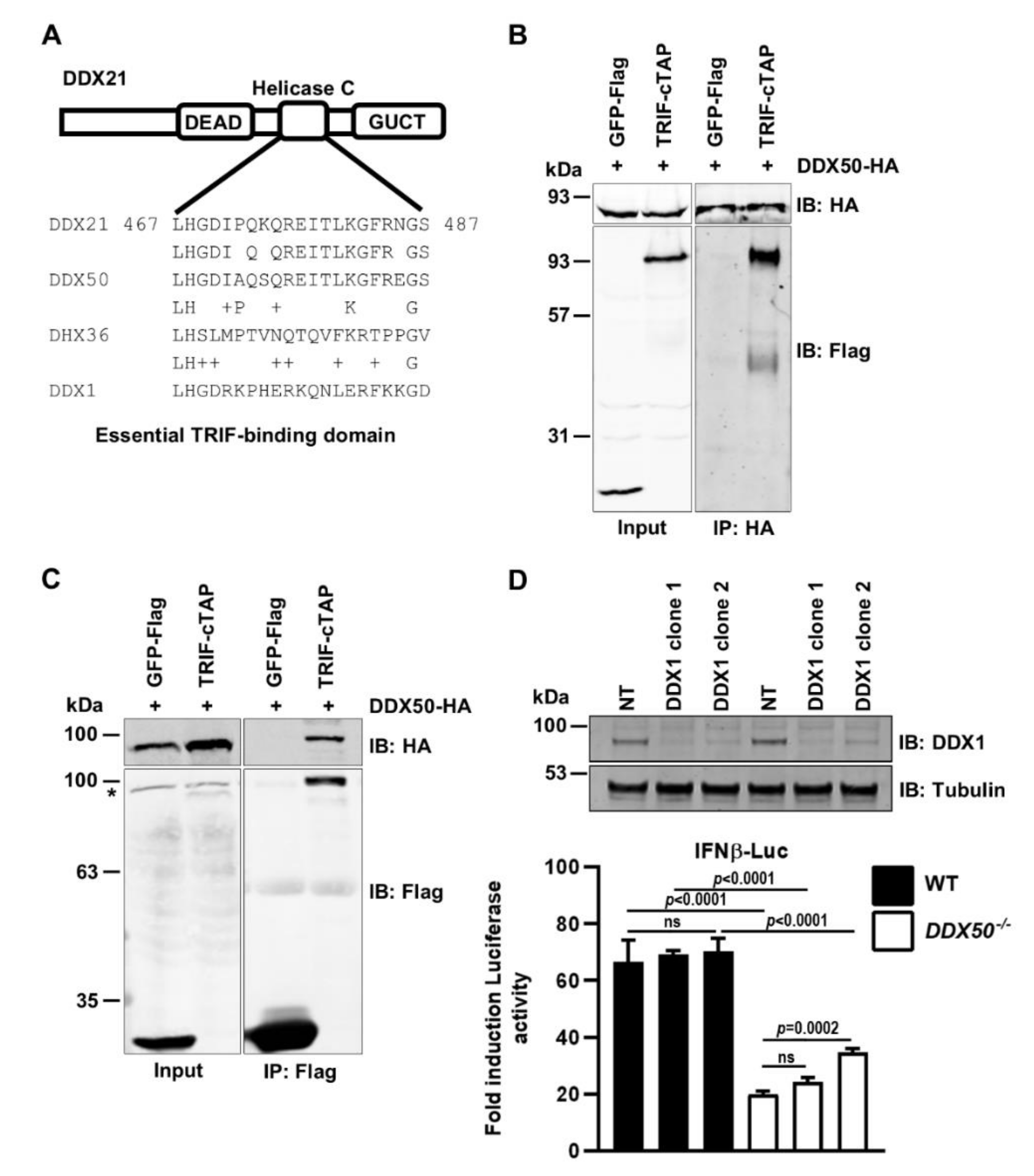
3.4. DDX50 Co-Immunoprecipitates with TRIF and Activates Signal Transduction Independently of the DDX1-DDX21-DHX36 Complex
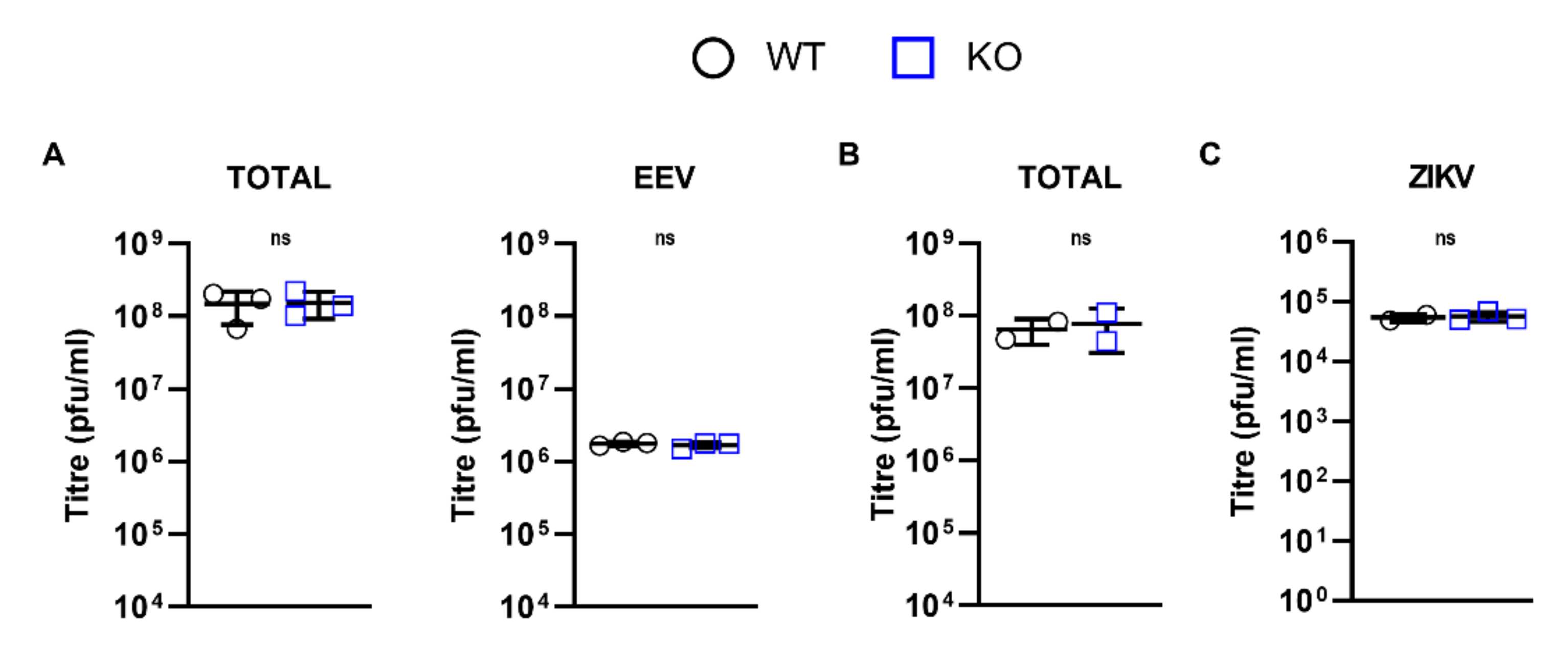
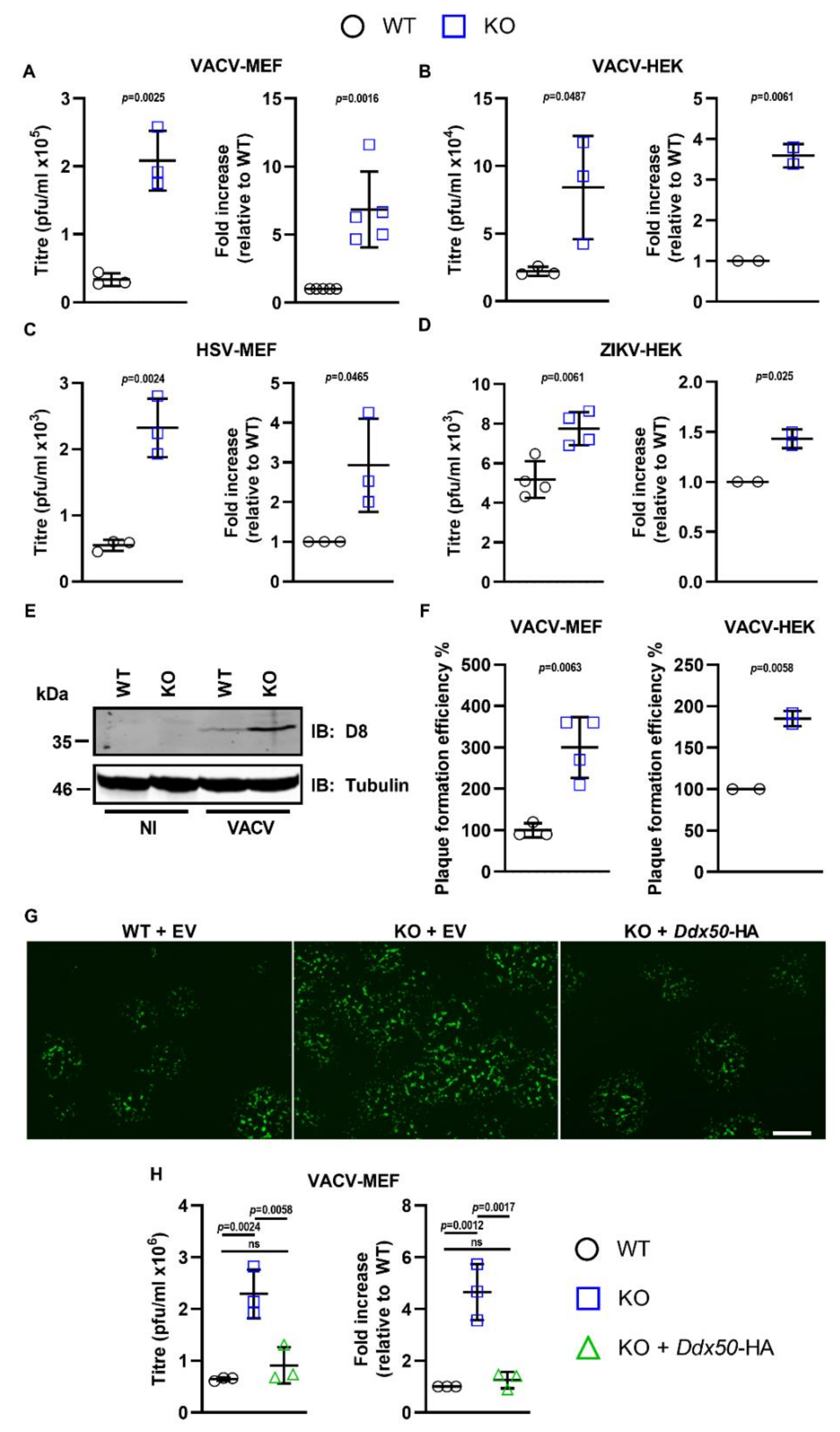
3.5. DDX50 Is a Viral Restriction Factor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Boxx, G.M.; Cheng, G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 2016, 19, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral Immunity via RIG-I-Mediated Recognition of RNA Bearing 5′-Diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of Cytosolic DNA Activates an IRF3-Dependent Innate Immune Response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullam, A.; Schroder, M. DExD/H-box RNA Helicases as Mediators of Anti-Viral Innate Immunity and Essential Host Factors for Viral Replication. Biochim. Biophys. Acta 2013, 1829, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, N.; Mitoma, H.; Kim, T.; Hanabuchi, S.; Liu, Y.-J. Helicase Proteins DHX29 and RIG-I Cosense Cytosolic Nucleic Acids in the Human Airway System. Proc. Natl. Acad. Sci. USA 2014, 111, 7747–7752. [Google Scholar] [CrossRef] [Green Version]
- De Nardo, D. Activation of the Innate Immune Receptors: Guardians of the Micro Galaxy: Activation and Functions of the Innate Immune Receptors. Adv. Exp. Med. Biol. 2017, 1024, 1–35. [Google Scholar] [CrossRef]
- Schafer, S.L.; Lin, R.; Moore, P.A.; Hiscott, J.; Pitha, P.M. Regulation of Type I Interferon Gene Expression by Interferon Regulatory Factor-3. J. Biol. Chem. 1998, 273, 2714–2720. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, T.; Sato, M.; Hata, N.; Asagiri, M.; Suemori, H.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Gene Induction Pathways Mediated by Distinct IRFs during Viral Infection. Biochem. Biophys. Res. Commun. 2001, 283, 1150–1156. [Google Scholar] [CrossRef]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.-K.; Krug, A.; Hopfner, K.-P. The C-Terminal Regulatory Domain Is the RNA 5′-Triphosphate Sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.-W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 Ring-Finger E3 Ubiquitin Ligase Is Essential for RIG-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-Methylation Provides a Molecular Signature for the Distinction of Self and Non-Self mRNA Dependent on the RNA Sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. DDX60, a DEXD/H Box Helicase, Is a Novel Antiviral Factor Promoting RIG-I-Like Receptor-Mediated Signaling. Mol. Cell. Biol. 2011, 31, 3802–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Miyashita, M.; Okamoto, M.; Morioka, Y.; Okabe, M.; Matsumoto, M.; Seya, T. DDX60 Is Involved in RIG-I-Dependent and Independent Antiviral Responses, and Its Function Is Attenuated by Virus-Induced EGFR Activation. Cell Rep. 2015, 11, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kim, T.; Bao, M.; Facchinetti, V.; Jung, S.Y.; Ghaffari, A.A.; Qin, J.; Cheng, G.; Liu, Y.-J. DDX1, DDX21, and DHX36 Helicases form a Complex with the Adaptor Molecule TRIF to Sense dsRNA in Dendritic Cells. Immunity 2011, 34, 866–878. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, H.; Hanabuchi, S.; Kim, T.; Bao, M.; Zhang, Z.; Sugimoto, N.; Liu, Y.-J. The DHX33 RNA Helicase Senses Cytosolic RNA and Activates the NLRP3 Inflammasome. Immunity 2013, 39, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-Box Helicase DDX3X Is a Critical Component of the TANK-Binding Kinase 1-Dependent Innate Immune Response. EMBO J. 2008, 27, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Taschuk, F.; Cherry, S. DEAD-Box Helicases: Sensors, Regulators, and Effectors for Antiviral Defense. Viruses 2020, 12, 181. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Tan, P.; Li, Y.; Lin, M.; Li, C.; Mao, J.; Cui, J.; Zhao, W.; Wang, H.Y.; Wang, R.-F. DHX29 Functions as an RNA Co-Sensor for MDA5-Mediated EMCV-Specific Antiviral Immunity. PLoS Pathog. 2018, 14, e1006886. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; van der Veen, A.G.; Chakravarty, P.; Lin, R.; Rogers, N.; Rehwinkel, J.; Deddouche, S.; Rosewell, I.; Hiscott, J.; Sousa, C.R.E. Mouse Superkiller-2-Like Helicase DDX60 Is Dispensable for Type I IFN Induction and Immunity to Multiple Viruses. Eur. J. Immunol. 2015, 45, 3386–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Lee, R.; Feng, Q.; Langereis, M.A.; Ter Horst, R.; Szklarczyk, R.; Netea, M.G.; Andeweg, A.C.; van Kuppeveld, F.J.M.; Huynen, M.A. Integrative Genomics-Based Discovery of Novel Regulators of the Innate Antiviral Response. PLoS Comput. Biol. 2015, 11, e1004553. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, S.; Paakkonen, K.; Koshiba, S.; Tochio, N.; Sato, M.; Kobayashi, N.; Harada, T.; Watanabe, S.; Muto, Y.; Guntert, P.; et al. Solution Structure of the GUCT Domain from Human RNA Helicase II/Gu Beta Reveals the RRM Fold, but Implausible RNA Interactions. Proteins 2009, 74, 133–144. [Google Scholar] [CrossRef]
- Valdez, B.C.; Perlaky, L.; Henning, D. Expression, Cellular Localization, and Enzymatic Activities of RNA Helicase II/Gu(Beta). Exp. Cell Res. 2002, 276, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Westermarck, J.; Weiss, C.; Saffrich, R.; Kast, J.; Musti, A.-M.; Wessely, M.; Ansorge, W.; Seraphin, B.; Wilm, M.; Valdez, B.C.; et al. The DEXD/H-box RNA Helicase RHII/Gu Is a Co-Factor for C-Jun-Activated Transcription. EMBO J. 2002, 21, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Carter, G.C.; Rodger, G.; Murphy, B.J.; Law, M.; Krauss, O.; Hollinshead, M.; Smith, G.L. Vaccinia Virus Cores Are Transported on Microtubules. J. Gen. Virol. 2003, 84, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified Vaccinia Virus Ankara Undergoes Limited Replication in Human Cells and Lacks Several Immunomodulatory Proteins: Implications for Use as a Human Vaccine. J. Gen. Virol. 1998, 79, 1159–1167. [Google Scholar] [CrossRef]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic Tubules Regulated by Rab GTPases 5 and 11 Are Used for Envelopment of Herpes Simplex Virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D.; Bell, A.J.; Lu, Y.; Orr, A. The Replication Defect of ICP0-Null Mutant Herpes Simplex Virus 1 Can Be Largely Complemented by the Combined Activities of Human Cytomegalovirus Proteins IE1 and pp71. J. Virol. 2013, 87, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Žusinaite, E.; Mahalingam, S.; Merits, A. Reverse Genetic System, Genetically Stable Reporter Viruses and Packaged Subgenomic Replicon Based on a Brazilian Zika Virus Isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallett, M.A.; Ren, H.; Zhang, R.-Y.; Scutts, S.R.; Gonzalez, L.; Zhu, Z.; Maluquer de Motes, C.; Smith, G.L. Vaccinia Virus BBK E3 Ligase Adaptor A55 Targets Importin-Dependent NF-kappaB Activation and Inhibits CD8(+) T-Cell Memory. J. Virol. 2019, 93, e00051-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallett, M.A.; Crepin, V.F.; Serafini, N.; Habibzay, M.; Kotik, O.; Sanchez-Garrido, J.; Di Santo, J.P.; Shenoy, A.R.; Berger, C.N.; Frankel, G. Bacterial Virulence Factor Inhibits Caspase-4/11 Activation in Intestinal Epithelial Cells. Mucosal Immunol. 2017, 10, 602–612. [Google Scholar] [CrossRef]
- Pallett, M.A.; Berger, C.N.; Pearson, J.S.; Hartland, E.L.; Frankel, G. The Type III Secretion Effector NleF of Enteropathogenic Escherichia Coli Activates NF-kappaB Early during Infection. Infect. Immun. 2014, 82, 4878–4888. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia Virus Protein C6 Is a Virulence Factor That Binds TBK-1 Adaptor Proteins and Inhibits Activation of IRF3 and IRF7. PLoS Pathog. 2011, 7, e1002247. [Google Scholar] [CrossRef]
- Parkinson, J.E.; Smith, G.L. Vaccinia Virus Gene A36R Encodes a M(r) 43-50 K Protein on the Surface of Extracellular Enveloped Virus. Virology 1994, 204, 376–390. [Google Scholar] [CrossRef]
- Lehmann, M.H.; Kastenmuller, W.; Kandemir, J.D.; Brandt, F.; Suezer, Y.; Sutter, G. Modified Vaccinia Virus Ankara Triggers Chemotaxis of Monocytes and Early Respiratory Immigration of Leukocytes by Induction of CCL2 Expression. J. Virol. 2009, 83, 2540–2552. [Google Scholar] [CrossRef] [Green Version]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The Formation and Function of Extracellular Enveloped Vaccinia Virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [CrossRef]
- Roberts, K.L.; Smith, G.L. Vaccinia Virus Morphogenesis and Dissemination. Trends Microbiol. 2008, 16, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.M.; Bidet, K.; Yinglin, A.; Ler, S.G.; Hogue, K.; Blackstock, W.; Gunaratne, J.; Garcia-Blanco, M.A. Quantitative Mass Spectrometry of DENV-2 RNA-Interacting Proteins Reveals that the DEAD-Box RNA Helicase DDX6 Binds the DB1 and DB2 3′ UTR Structures. RNA Biol. 2011, 8, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Feng, T.; Pan, W.; Shi, X.; Dai, J. DEAD-Box RNA Helicase DDX3X Inhibits DENV Replication via Regulating Type One Interferon Pathway. Biochem. Biophys. Res. Commun. 2015, 456, 327–332. [Google Scholar] [CrossRef]
- Han, P.; Ye, W.; Lv, X.; Ma, H.; Weng, D.; Dong, Y.; Cheng, L.; Chen, H.; Zhang, L.; Xu, Z.; et al. DDX50 Inhibits the Replication of Dengue Virus 2 by Upregulating IFN-Beta Production. Arch. Virol. 2017, 162, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Majumdar, T.; Kessler, P.; Ozhegov, E.; Zhang, Y.; Chattopadhyay, S.; Barik, S.; Sen, G.C. STING Requires the Adaptor TRIF to Trigger Innate Immune Responses to Microbial Infection. Cell Host Microbe 2016, 20, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.-P.; Gack, M.U. Viral Unmasking of Cellular 5S rRNA Pseudogene Transcripts Induces RIG-I-Mediated Immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.P.; Roberts, W.K.; Kerr, I.M. 2-5A Accumulates to High Levels in Interferon-Treated, Vaccinia Virus-Infected Cells in the Absence of Any Inhibition of Virus Replication. J. Virol. 1984, 50, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-W.; Katsafanas, G.C.; Liu, R.; Wyatt, L.S.; Moss, B. Poxvirus Decapping Enzymes Enhance Virulence by Preventing the Accumulation of dsRNA and the Induction of Innate Antiviral Responses. Cell Host Microbe 2015, 17, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Moss, B.; Smith, G.L. Poxviridae: The viruses and their replication. In Fields Virology, 7th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health: Philadelphia, PA, USA, 2021; Volume 2, pp. 573–613. [Google Scholar]
- Price, P.J.R.; Torres-Domínguez, L.E.; Brandmüller, C.; Sutter, G.; Lehmann, M.H. Modified Vaccinia Virus Ankara: Innate Immune Activation and Induction of Cellular Signalling. Vaccine 2013, 31, 4231–4234. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L Gene of Vaccinia Virus Encodes an Inhibitor of the Interferon-Induced, Double-Stranded RNA-Dependent Protein Kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, T.A.; Jacobs, B.L. Both Carboxy- and Amino-Terminal Domains of the Vaccinia Virus Interferon Resistance Gene, E3L, are Required for Pathogenesis in a Mouse Model. J. Virol. 2001, 75, 850–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallett, M.A.; Lu, Y.; Smith, G.L. DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation. Viruses 2022, 14, 316. https://doi.org/10.3390/v14020316
Pallett MA, Lu Y, Smith GL. DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation. Viruses. 2022; 14(2):316. https://doi.org/10.3390/v14020316
Chicago/Turabian StylePallett, Mitchell A., Yongxu Lu, and Geoffrey L. Smith. 2022. "DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation" Viruses 14, no. 2: 316. https://doi.org/10.3390/v14020316
APA StylePallett, M. A., Lu, Y., & Smith, G. L. (2022). DDX50 Is a Viral Restriction Factor That Enhances IRF3 Activation. Viruses, 14(2), 316. https://doi.org/10.3390/v14020316