
Human SUMOylation Pathway Is Critical for Influenza B Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Cloning of DNA Constructs
2.2. Cell Lines
2.3. Protein Expression and Purification
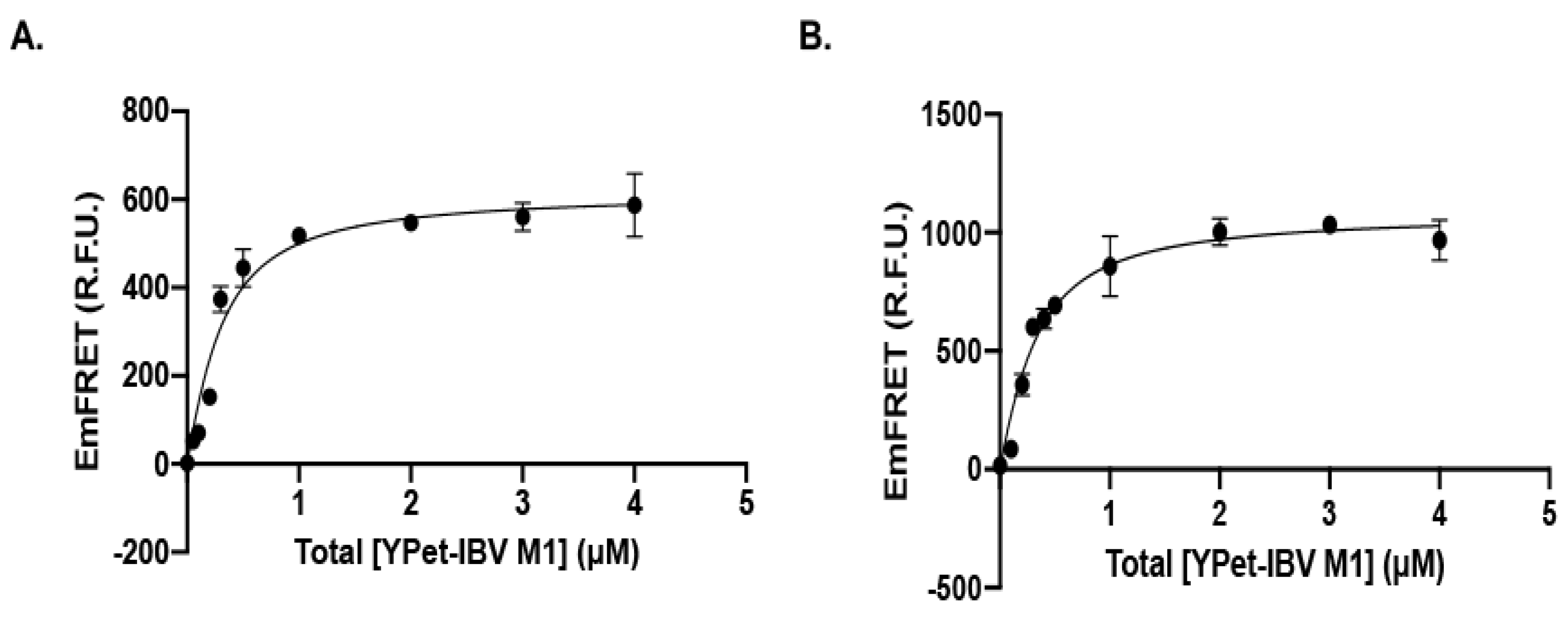
2.4. qFRET Determination of the Dissociation Constant (KD)
2.5. qFRET In Vitro SUMOylation Assay
2.6. EmFRET Analysis
2.7. Reconstitution of Wild-Type Influenza B Virus (B/Yamagata/16/1988) and M1 Mutant Influenza B Virus (B/Yamagata/16/1988)
2.8. Plaque Assay
2.9. Natural Red Assay
3. Results
3.1. Identification of the Human SUMOylation Pathway as a Critical Host Pathway for IBV’s Life Cycle
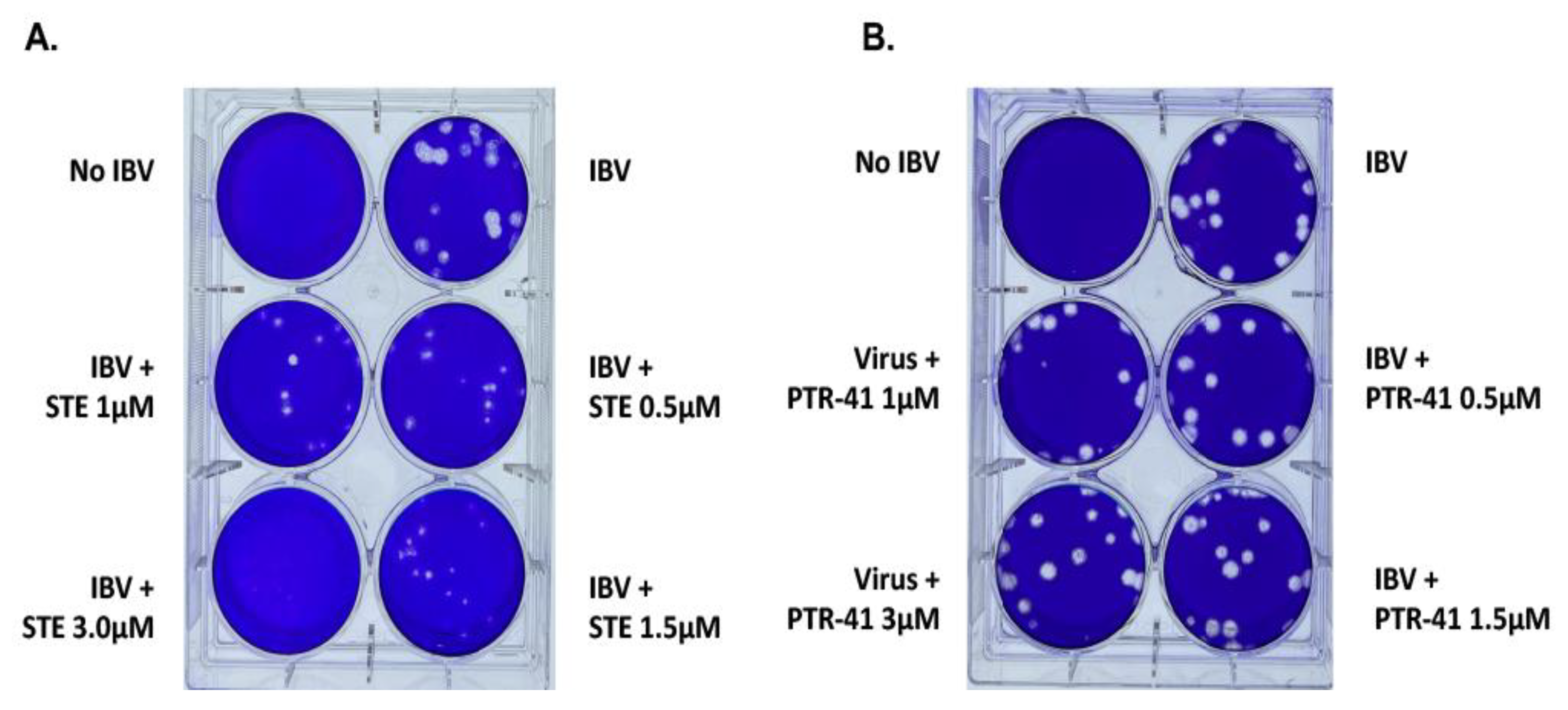
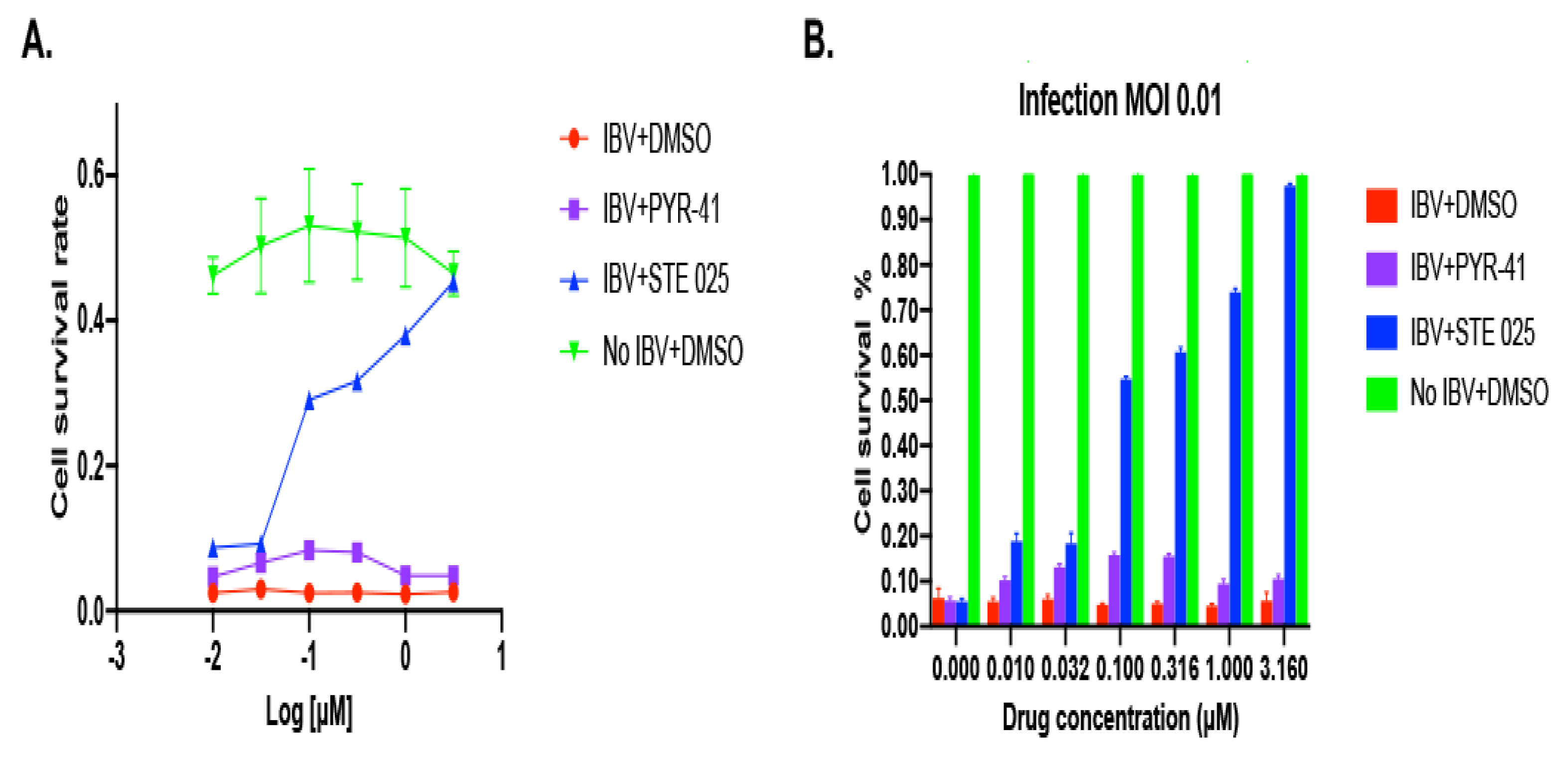
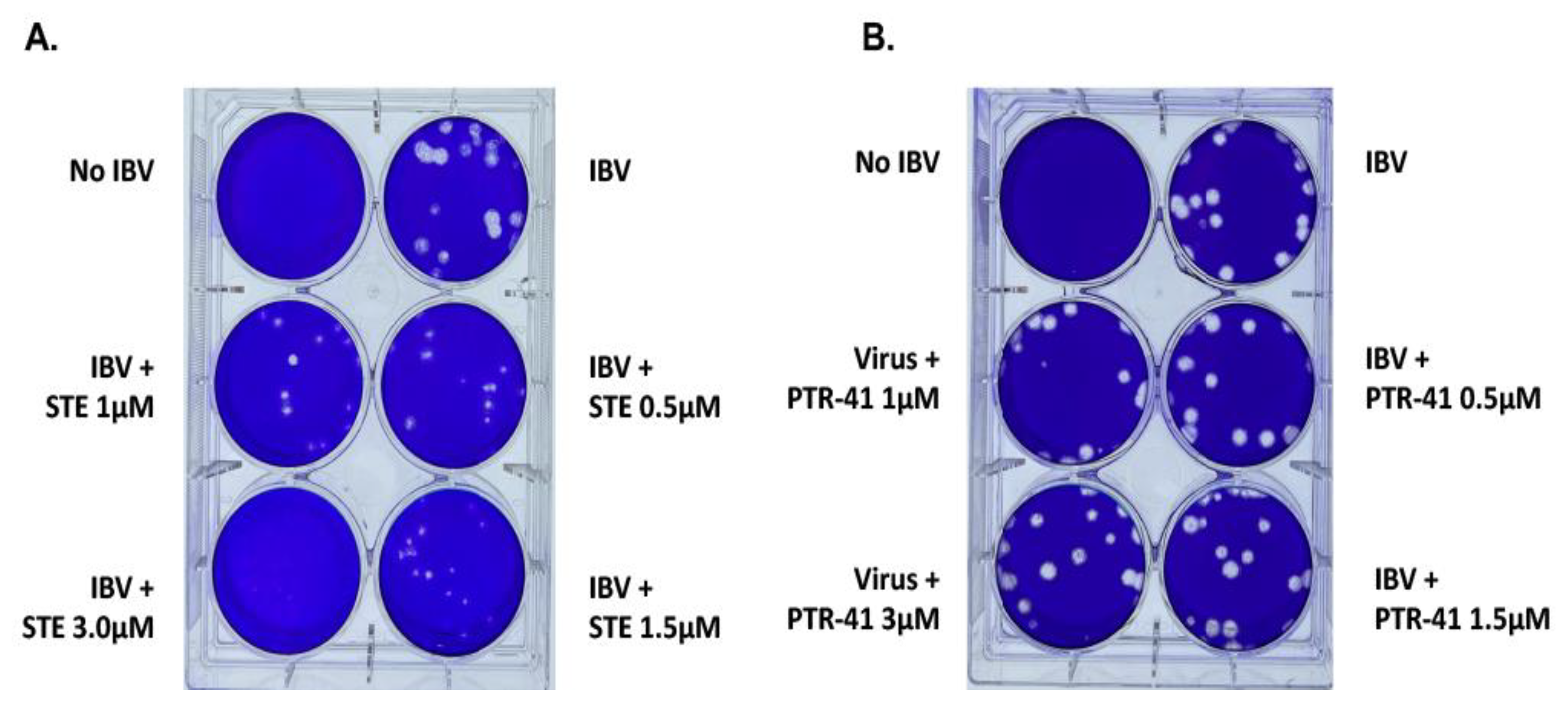
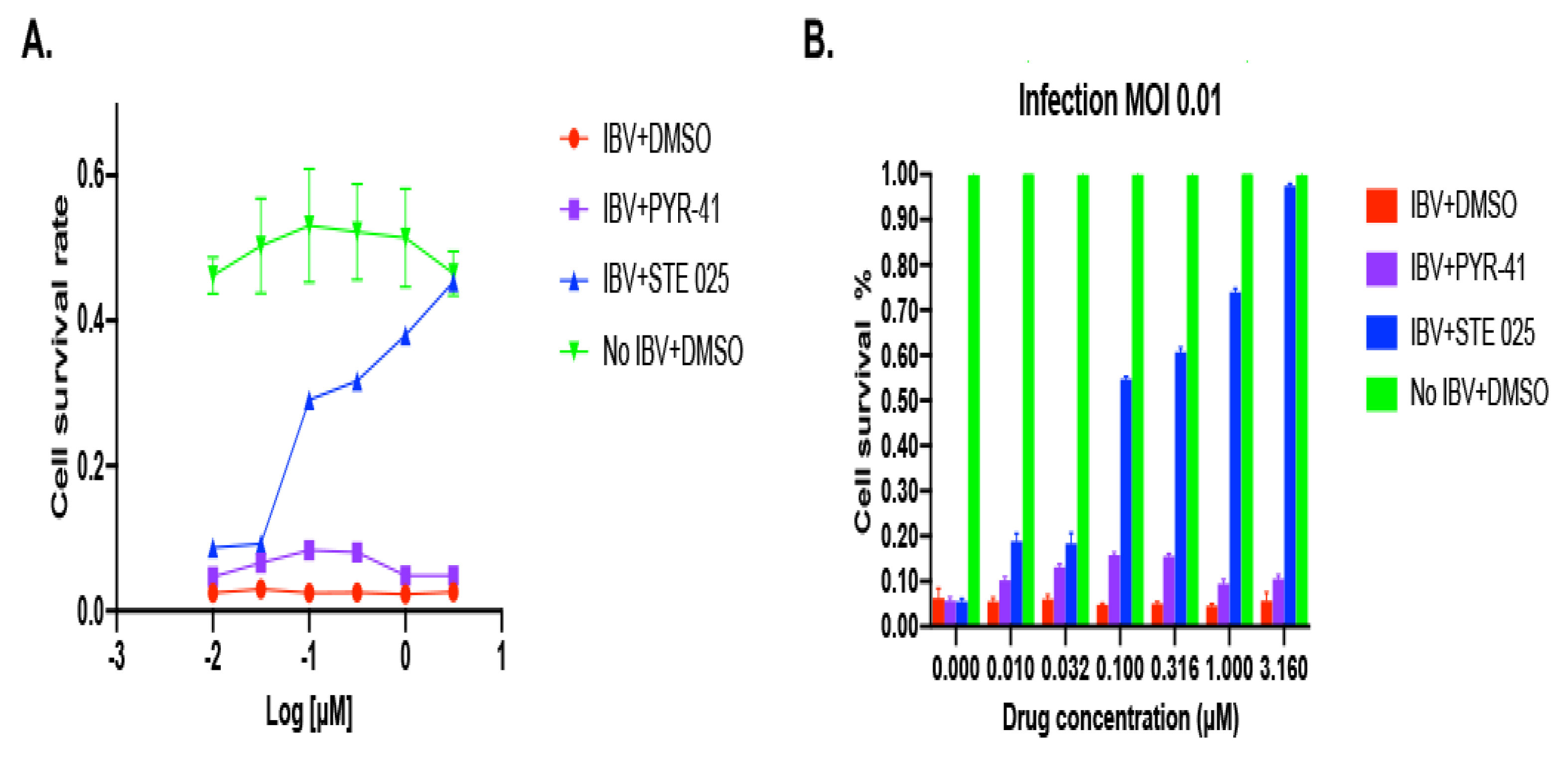
3.1.1. IBV Is Completely Inhibited by the SUMOylation Specific Inhibitor
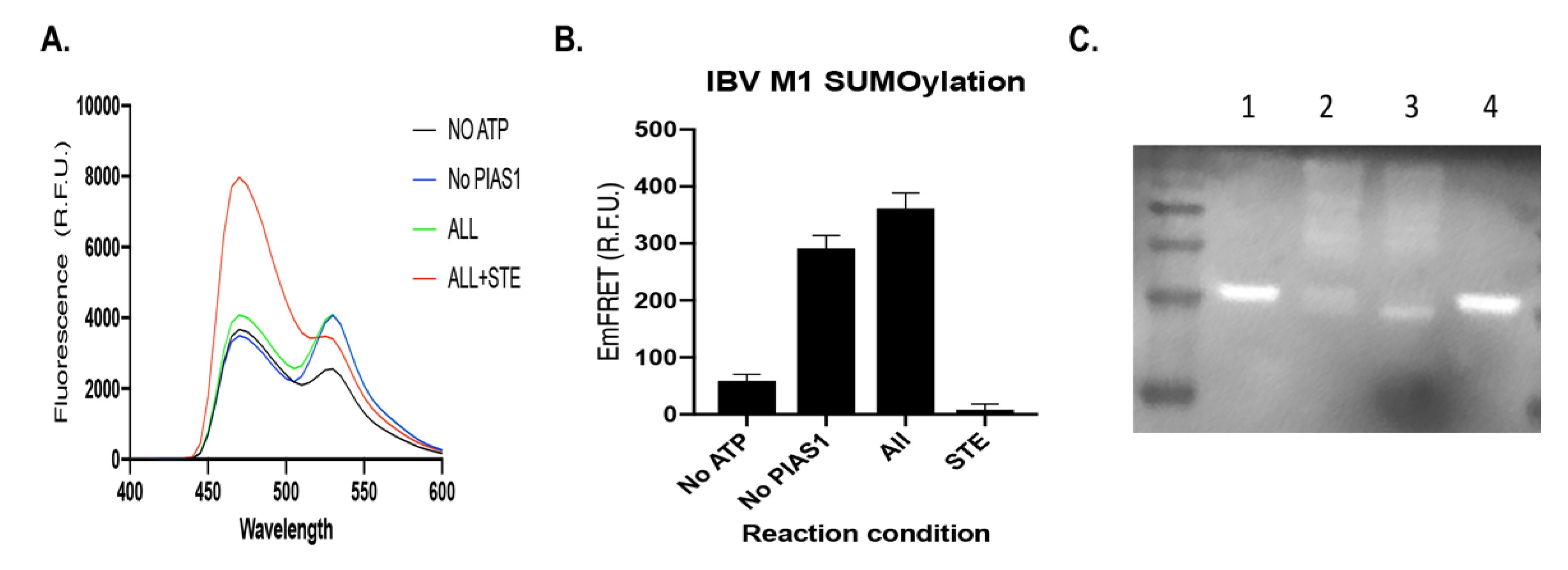
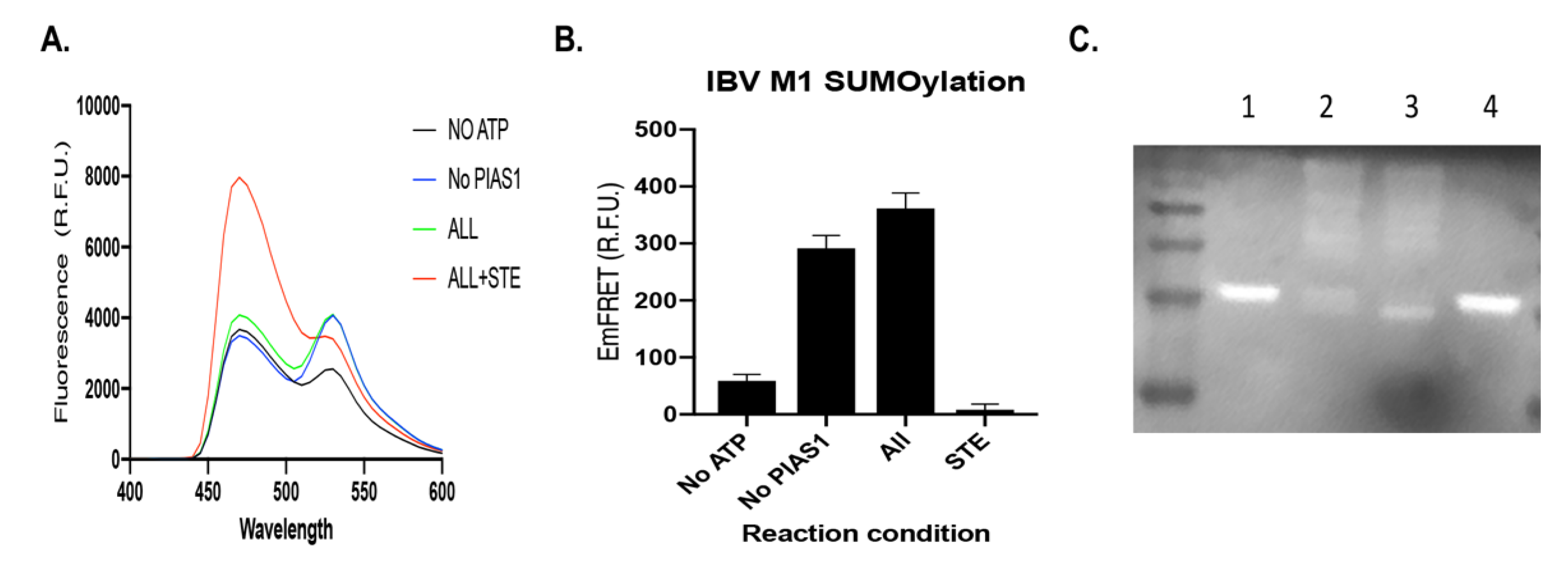
3.1.2. IBV M1 Protein as a Target of SUMOylation
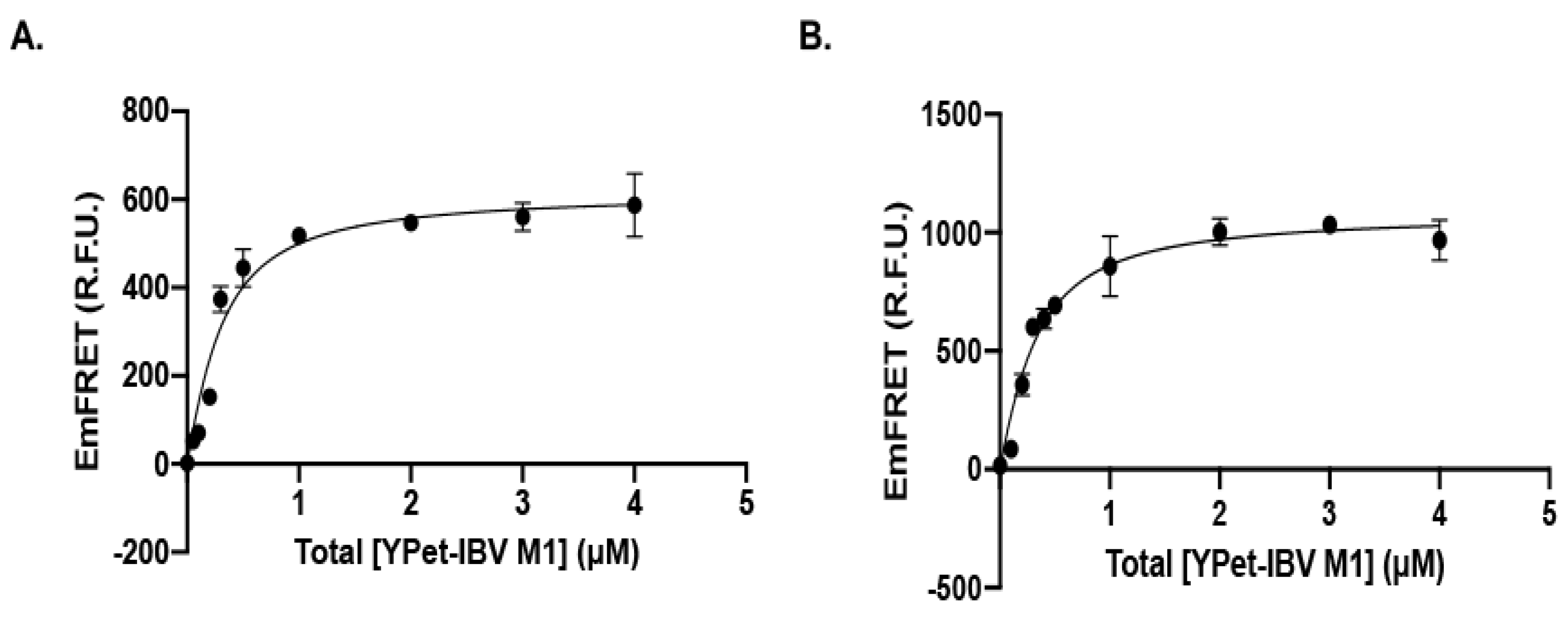
3.1.3. Recognition of IBV M1 Protein by the SUMOylation E2 and E3 with High Affinity
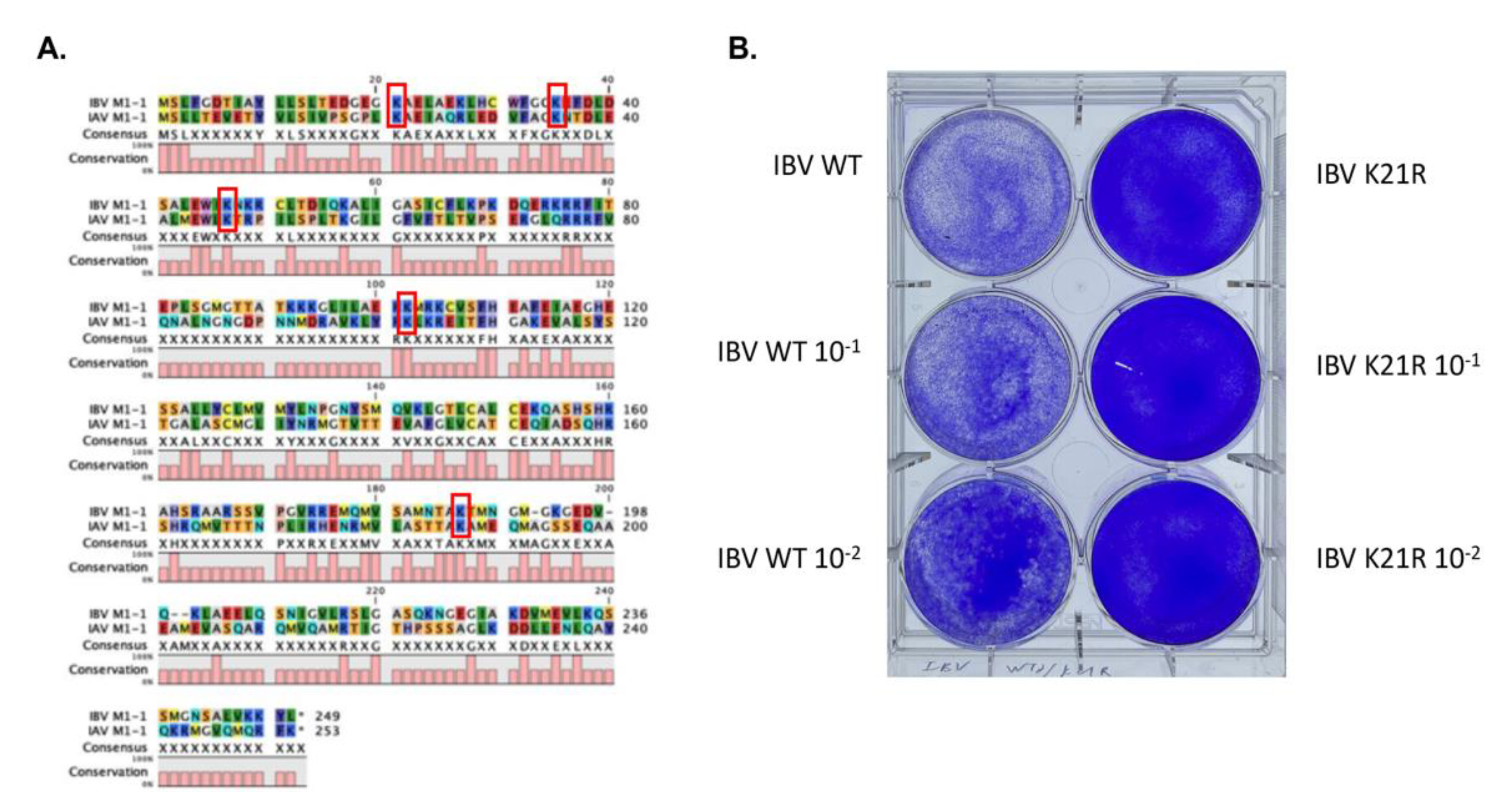
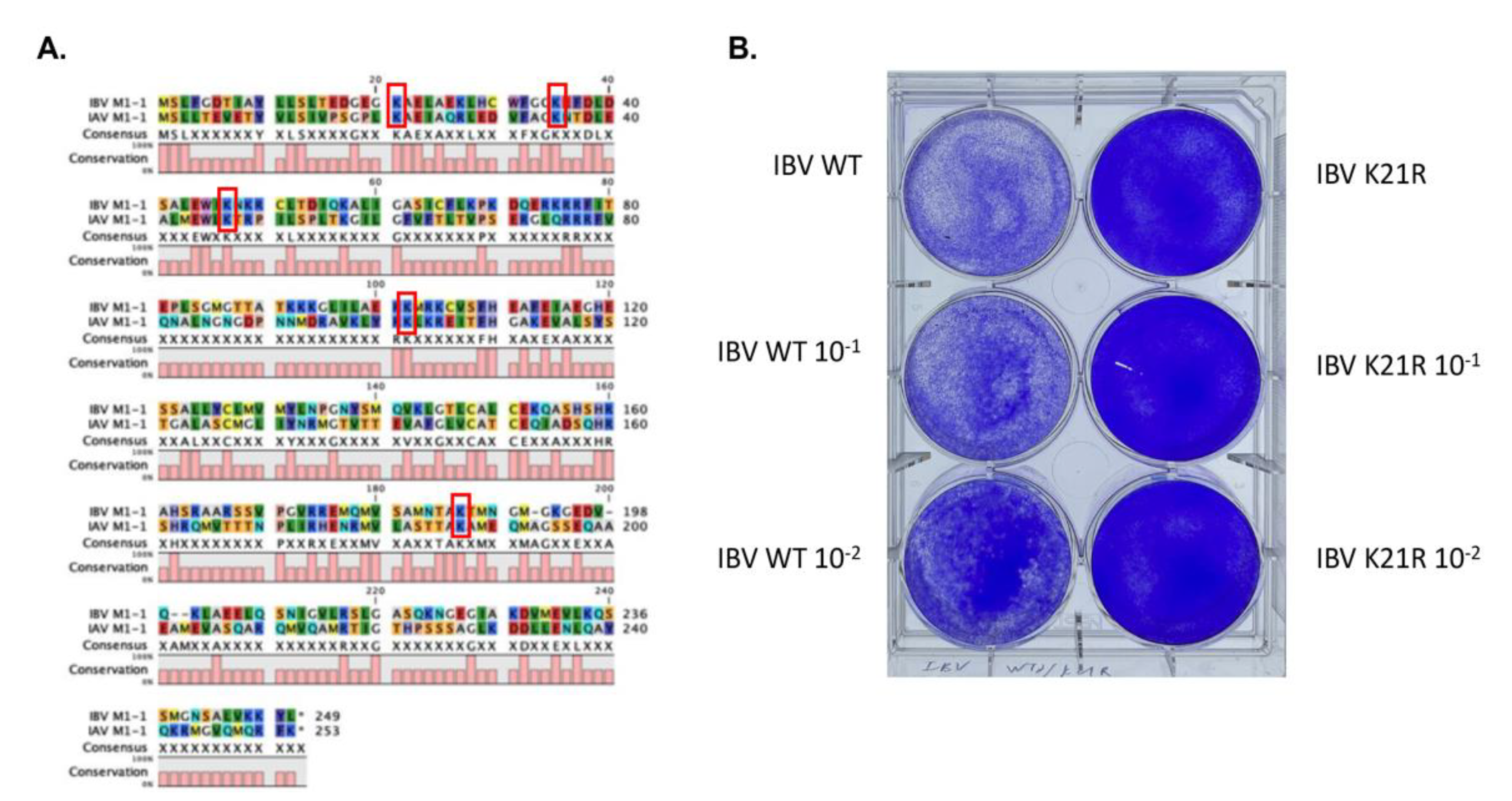
3.1.4. Identification of an Essential SUMOylation Site of the IBV M1 Protein
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Estimated Flu-Related Illnesses, Medical Visits, Hospitalizations, and Deaths in the United States—2017–2018 Flu Season. 2019. Available online: https://www.cdc.gov/flu/about/burden/2018-2019.html (accessed on 27 December 2021).
- Putri, W.; Muscatello, D.J.; Stockwell, M.S.; Newall, A.T. Economic burden of seasonal influenza in the United States. Vaccine 2018, 36, 3960–3966. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Yan, S.; Weycker, D.; Sokolowski, S. US healthcare costs attributable to type A and type B influenza. Hum. Vaccines Immunother. 2017, 13, 2041–2047. [Google Scholar] [CrossRef] [Green Version]
- Mosnier, A.; Caini, S.; Daviaud, I.; Nauleau, E.; Bui, T.T.; Debost, E.; Bedouret, B.; Agius, G.; van der Werf, S.; Lina, B.; et al. Clinical Characteristics Are Similar across Type A and B Influenza Virus Infections. PLoS ONE 2015, 10, e0136186. [Google Scholar] [CrossRef] [PubMed]
- Bhat, Y.R. Influenza B infections in children: A review. World J. Clin. Pediatr. 2020, 9, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Vaudry, W.; Moore, D.; Bettinger, J.A.; Halperin, S.A.; Scheifele, D.W.; Jadvji, T.; Lee, L.; Mersereau, T.; members of the Canadian Immunization Monitoring Program, A. Hospitalization for Influenza A Versus B. Pediatrics 2016, 138, e20154643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.; Moyes, J.; Tempia, S.; Groom, M.; Walaza, S.; Pretorius, M.; Dawood, H.; Chhagan, M.; Haffejee, S.; Variava, E.; et al. Severe influenza-associated respiratory infection in high HIV prevalence setting, South Africa, 2009–2011. Emerg. Infect. Dis. 2013, 19, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Influenza-Associated Pediatric Deaths—United States, September 2010–August 2011. MMWR Morb. Mortal. Wkly. Rep. 2011, 60, 1233–1238. [Google Scholar]
- Centers for Disease Control and Prevention. Quadrivalent Influenza Vaccine. Available online: https://www.cdc.gov/flu/prevent/quadrivalent.htm (accessed on 27 December 2021).
- Centers for Disease Control and Prevention. US Flu VE Data for 2019–2020. Available online: https://www.cdc.gov/flu/about/burden/2019-2020.html (accessed on 27 December 2021).
- Monto, A.S.; McKimm-Breschkin, J.L.; Macken, C.; Hampson, A.W.; Hay, A.; Klimov, A.; Tashiro, M.; Webster, R.G.; Aymard, M.; Hayden, F.G.; et al. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob. Agents Chemother. 2006, 50, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Sheu, T.G.; Deyde, V.M.; Okomo-Adhiambo, M.; Garten, R.J.; Xu, X.; Bright, R.A.; Butler, E.N.; Wallis, T.R.; Klimov, A.I.; Gubareva, L.V. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 2008, 52, 3284–3292. [Google Scholar] [CrossRef] [Green Version]
- Dharan, N.J.; Gubareva, L.V.; Meyer, J.J.; Okomo-Adhiambo, M.; McClinton, R.C.; Marshall, S.A.; St George, K.; Epperson, S.; Brammer, L.; Klimov, A.I.; et al. Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA 2009, 301, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liao, H.; Liu, Y.; Liu, L.; Wang, F.; Song, H.; Cheng, J.; Liu, X.; Xu, D. Drug-Resistant and Genetic Evolutionary Analysis of Influenza Virus from Patients During the 2013 and 2014 Influenza Season in Beijing. Microb. Drug Resist. 2017, 23, 253–260. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Mizuta, K.; Aoki, Y.; Suto, A.; Abiko, C.; Sanjoh, K.; Sugawara, K.; Takashita, E.; Itagaki, T.; Katsushima, Y.; et al. A two-year survey of the oseltamivir-resistant influenza A(H1N1) virus in Yamagata, Japan and the clinical effectiveness of oseltamivir and zanamivir. Virol. J. 2010, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, S.; Sugaya, N.; Ito, M.; Yamazaki, M.; Ichikawa, M.; Kimura, K.; Kiso, M.; Shimizu, H.; Kawakami, C.; Koike, K.; et al. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA 2007, 297, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Abed, Y.; Fage, C.; Lague, P.; Carbonneau, J.; Papenburg, J.; Vinh, D.C.; Boivin, G. Reduced Susceptibility to Neuraminidase Inhibitors in Influenza B Isolate, Canada. Emerg. Infect. Dis. 2019, 25, 838–840. [Google Scholar] [CrossRef] [Green Version]
- Kawai, N.; Ikematsu, H.; Iwaki, N.; Kawashima, T.; Maeda, T.; Mitsuoka, S.; Kondou, K.; Satoh, I.; Miyachi, K.; Yamaga, S.; et al. Longer virus shedding in influenza B than in influenza A among outpatients treated with oseltamivir. J. Infect. 2007, 55, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.; Jones, M.A.; Doshi, P.; Del Mar, C.B.; Hama, R.; Thompson, M.J.; Spencer, E.A.; Onakpoya, I.; Mahtani, K.R.; Nunan, D.; et al. Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane Database Syst. Rev. 2014, 2014, CD008965. [Google Scholar] [CrossRef] [PubMed]
- Ison, M.G.; Portsmouth, S.; Yoshida, Y.; Shishido, T.; Mitchener, M.; Tsuchiya, K.; Uehara, T.; Hayden, F.G. Early treatment with baloxavir marboxil in high-risk adolescent and adult outpatients with uncomplicated influenza (CAPSTONE-2): A randomised, placebo-controlled, phase 3 trial. Lancet Infect. Dis. 2020, 20, 1204–1214. [Google Scholar] [CrossRef]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Mink, S.; Wong, K.A.; Stein, N.; Getman, C.; Dempsey, P.W.; Wu, H.; Shuai, K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004, 5, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Santos, A.; Rosas, J.M.; Ortiz-Guzman, J.; Rosas-Acosta, G. Influenza A virus interacts extensively with the cellular SUMOylation system during infection. Virus Res. 2011, 158, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, P.; Schreiner, S.; Dobner, T. Human pathogens and the host cell SUMOylation system. J. Virol. 2012, 86, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggio, R.; Chiocca, S. Viruses and sumoylation: Recent highlights. Curr. Opin. Microbiol. 2006, 9, 430–436. [Google Scholar] [CrossRef]
- Xu, K.; Klenk, C.; Liu, B.; Keiner, B.; Cheng, J.; Zheng, B.J.; Li, L.; Han, Q.; Wang, C.; Li, T.; et al. Modification of nonstructural protein 1 of influenza A virus by SUMO1. J. Virol. 2011, 85, 1086–1098. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Jeng, K.S.; Lai, M.M. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J. Virol. 2011, 85, 6618–6628. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Chang, C.; Li, L.; Klenk, C.; Cheng, J.; Chen, Y.; Xia, N.; Shu, Y.; Chen, Z.; Gabriel, G.; et al. Sumoylation of influenza A virus nucleoprotein is essential for intracellular trafficking and virus growth. J. Virol. 2014, 88, 9379–9390. [Google Scholar] [CrossRef] [Green Version]
- Way, G.; Xiong, Z.; Wang, G.; Dai, H.; Zheng, S.; Garcia-Sastre, A.; Liao, J. A novel SUMOylation site in the influenza a virus NS1 protein identified with a highly sensitive FRET assay. J. Biotechnol. 2020, 323, 121–127. [Google Scholar] [CrossRef]
- Furuse, Y.; Suzuki, A.; Kamigaki, T.; Oshitani, H. Evolution of the M gene of the influenza A virus in different host species: Large-scale sequence analysis. Virol. J. 2009, 6, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Gorman, O.T.; Kawaoka, Y.; Bean, W.J.; Webster, R.G. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J. Virol. 1991, 65, 5491–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelle, K.; Cobey, S.; Grenfell, B.; Pascual, M. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 2006, 314, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Choo, J.A.; Liu, J.; Toh, X.; Grotenbreg, G.M.; Ren, E.C. The immunodominant influenza A virus M158-66 cytotoxic T lymphocyte epitope exhibits degenerate class I major histocompatibility complex restriction in humans. J. Virol. 2014, 88, 10613–10623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Sandt, C.E.; Kreijtz, J.H.; Geelhoed-Mieras, M.M.; Nieuwkoop, N.J.; Spronken, M.I.; van de Vijver, D.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Differential Recognition of Influenza A Viruses by M158-66 Epitope-Specific CD8+ T Cells Is Determined by Extraepitopic Amino Acid Residues. J. Virol. 2016, 90, 1009–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Liao, J. Systematic determinations of SUMOylation activation intermediates and dynamics by a sensitive and quantitative FRET assay. Mol. Biosyst. 2012, 8, 1723–1729. [Google Scholar] [CrossRef]
- Malik-Chaudhry, H.K.; Saavedra, A.; Liao, J. A linker strategy for trans-FRET assay to determine activation intermediate of NEDDylation cascade. Biotechnol. Bioeng. 2014, 111, 1288–1295. [Google Scholar] [CrossRef]
- Fodor, E.; Devenish, L.; Engelhardt, O.G.; Palese, P.; Brownlee, G.G.; Garcia-Sastre, A. Rescue of influenza A virus from recombinant DNA. J. Virol. 1999, 73, 9679–9682. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET Assay into Quantitative and High-throughput Screening Technology Platforms for Protein-Protein Interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011, 12, 4027–4052. [Google Scholar] [CrossRef] [Green Version]
- Kellam, P. Attacking pathogens through their hosts. Genome Biol. 2006, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- De Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; Andre, P.; Lotteau, V. Virus-host interactomics: New insights and opportunities for antiviral drug discovery. Genome Med. 2014, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kitagaki, J.; Dai, R.M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Madahar, V.; Dang, R.; Jiang, L. Quantitative FRET (qFRET) Technology for the Determination of Protein-Protein Interaction Affinity in Solution. Molecules 2021, 26, 6339. [Google Scholar] [CrossRef]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarge, K.D.; Park-Sarge, O.K. SUMO and its role in human diseases. Int. Rev. Cell Mol. Biol. 2011, 288, 167–183. [Google Scholar]
- Reverter, D.; Lima, C.D. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 2005, 435, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Yunus, A.A.; Lima, C.D. Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Mol. Cell 2009, 35, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation—A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef]
- Song, Y.; Rodgers, V.G.; Schultz, J.S.; Liao, J. Protein interaction affinity determination by quantitative FRET technology. Biotechnol. Bioeng. 2012, 109, 2875–2883. [Google Scholar] [CrossRef]
- Lowrey, A.J.; Cramblet, W.; Bentz, G. Viral manipulation of the cellular sumoylation machinery. Cell Commun. Signal. 2017, 15, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, O.; Kessler, N.; Lina, B. Sensitivity of influenza viruses to zanamivir and oseltamivir: A study performed on viruses circulating in France prior to the introduction of neuraminidase inhibitors in clinical practice. Antivir. Res. 2005, 68, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liao, J.; Rao, X.; Kushner, S.A.; Chung, C.D.; Chang, D.D.; Shuai, K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 1998, 95, 10626–10631. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Fu, Y.; Shuai, K. Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of activated signal transducer and activator of transcription (STAT) 1 (PIAS1) in cytokine-induced PIAS1-Stat1 interaction. Proc. Natl. Acad. Sci. USA 2000, 97, 5267–5272. [Google Scholar] [CrossRef] [Green Version]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Baz-Martinez, M.; El Motiam, A.; Ruibal, P.; Condezo, G.N.; de la Cruz-Herrera, C.F.; Lang, V.; Collado, M.; San Martin, C.; Rodriguez, M.S.; Munoz-Fontela, C.; et al. Regulation of Ebola virus VP40 matrix protein by SUMO. Sci. Rep. 2016, 6, 37258. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [CrossRef] [Green Version]
- Bentz, G.L.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J. Virol. 2012, 86, 12251–12261. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 Proteome during HSV-1 Infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Li, C.; Luo, F.; Liu, C.; Xiong, N.; Xu, Z.; Zhang, W.; Yang, M.; Wang, Y.; Liu, D.; Yu, C.; et al. Effect of a genetically engineered interferon-alpha versus traditional interferon-alpha in the treatment of moderate-to-severe COVID-19: A randomised clinical trial. Ann. Med. 2021, 53, 391–401. [Google Scholar] [CrossRef]
- Domingues, P.; Golebiowski, F.; Tatham, M.H.; Lopes, A.M.; Taggart, A.; Hay, R.T.; Hale, B.G. Global Reprogramming of Host SUMOylation during Influenza Virus Infection. Cell Rep. 2015, 13, 1467–1480. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [Green Version]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Korona, D.A.; Lecompte, K.G.; Pursell, Z.F. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2011, 39, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Way, G.; Madahar, V. Target Virus or Target Ourselves for COVID-19 Drugs Discovery?-Lessons learned from anti-influenzas virus therapies. Med. Drug Discov. 2020, 100037. [Google Scholar] [CrossRef]
- Van de Wakker, S.I.; Fischer, M.J.E.; Oosting, R.S. New drug-strategies to tackle viral-host interactions for the treatment of influenza virus infections. Eur. J. Pharmacol. 2017, 809, 178–190. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- Liao, J. Protein Inhibitors of Activated Stat. Ph.D. Thesis, University of California, Los Angeles, CA, USA, 1999. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, R.; Rodgers, V.G.J.; García-Sastre, A.; Liao, J. Human SUMOylation Pathway Is Critical for Influenza B Virus. Viruses 2022, 14, 314. https://doi.org/10.3390/v14020314
Dang R, Rodgers VGJ, García-Sastre A, Liao J. Human SUMOylation Pathway Is Critical for Influenza B Virus. Viruses. 2022; 14(2):314. https://doi.org/10.3390/v14020314
Chicago/Turabian StyleDang, Runrui, Victor G. J. Rodgers, Adolfo García-Sastre, and Jiayu Liao. 2022. "Human SUMOylation Pathway Is Critical for Influenza B Virus" Viruses 14, no. 2: 314. https://doi.org/10.3390/v14020314
APA StyleDang, R., Rodgers, V. G. J., García-Sastre, A., & Liao, J. (2022). Human SUMOylation Pathway Is Critical for Influenza B Virus. Viruses, 14(2), 314. https://doi.org/10.3390/v14020314