
Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Iwi Consultation and Animal Ethics Approval
2.2. Fish Gill Meta-Transcriptomic Sequencing
2.3. Virome Assembly and Virus Identification
2.4. Virus Phylogenetic Analysis
2.5. Ecological Analysis of Virome Alpha and Beta Diversity
2.6. Viral Nomenclature
3. Results
3.1. Novel Fish Viruses
3.1.1. Astroviridae
3.1.2. Orthomyxoviridae
3.1.3. Paramyxoviridae
3.1.4. Rhabdoviridae
3.1.5. Totiviridae
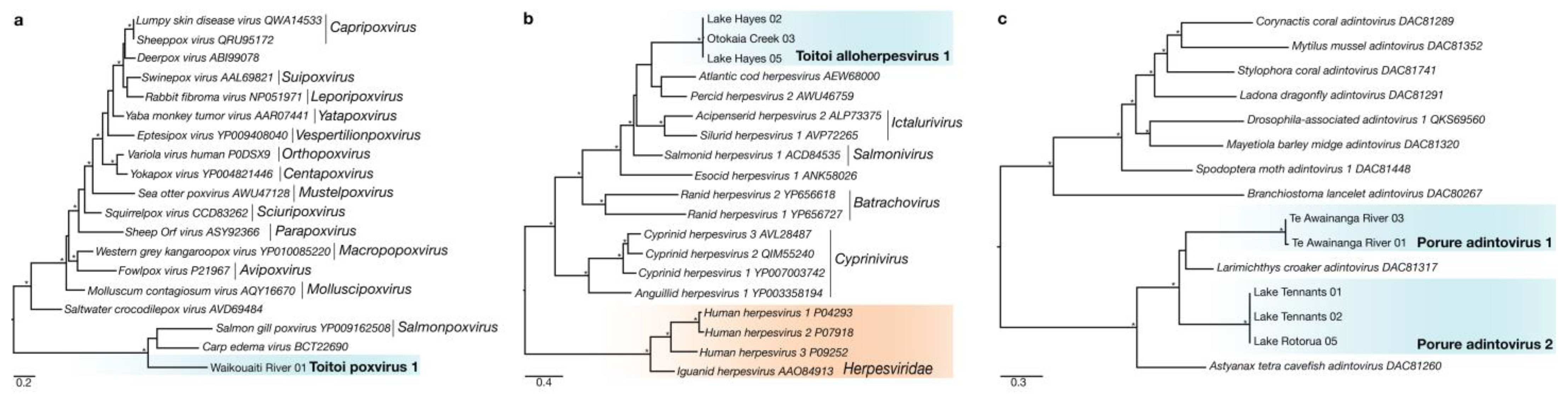
3.1.6. Poxviridae
3.1.7. Alloherpesviridae
3.1.8. Adintoviridae
3.2. Viral Abundance and Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodrich, E.S. The Origin of Land Vertebrates. Nature 1924, 114, 935–936. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Duchêne, S.; Holmes, E.C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLOS Pathog. 2017, 13, e1006215. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.G.; Bradshaw, J.D. The break-up of a long-term relationship: The Cretaceous separation of New Zealand from Gondwana. Gondwana Res. 2004, 7, 273–286. [Google Scholar] [CrossRef]
- Cooper, R.A.; Millener, P.R. The New Zealand biota: Historical background and new research. Trends Ecol. Evol. 1993, 8, 429–433. [Google Scholar] [CrossRef]
- Worthy, T.H.; Tennyson, A.J.D.; Archer, M.; Musser, A.M.; Hand, S.J.; Jones, C.; Douglas, B.J.; McNamara, J.A.; Beck, R.M.D. Miocene mammal reveals a Mesozoic ghost lineage on insular New Zealand, southwest Pacific. Proc. Natl. Acad. Sci. USA 2006, 103, 19419–19423. [Google Scholar] [CrossRef]
- McDowall, R.M. Extinction and endemism in New Zealand land Birds. Tuatara 1969, 17, 1–12. [Google Scholar]
- Chapple, D.G. New Zealand Lizards; Chapple, D.G., Ed.; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-41672-4. [Google Scholar]
- Allibone, R.; David, B.; Hitchmough, R.; Jellyman, D.; Ling, N.; Ravenscroft, P.; Waters, J. Conservation status of New Zealand freshwater fish, 2009. N. Z. J. Mar. Freshw. Res. 2010, 44, 271–287. [Google Scholar] [CrossRef]
- Taylor-Smith, B.; Morgan-Richards, M.; Trewick, S.A. Patterns of regional endemism among New Zealand invertebrates. N. Z. J. Zool. 2020, 47, 1–19. [Google Scholar] [CrossRef]
- Ingram, T.; Dutoit, L.; Mikheev, P.; Khan, S.; Schallenberg, M. Phenotypic, ecological, and genomic variation in common bully (Gobiomorphus cotidianus) populations along depth gradients in New Zealand’s southern great lakes. Can. J. Fish. Aquat. Sci. 2020, 77, 1678–1687. [Google Scholar] [CrossRef]
- McDowall, R.M. Crying wolf, crying foul, or crying shame: Alien salmonids and a biodiversity crisis in the southern cool-temperate galaxioid fishes? Rev. Fish Biol. Fish. 2006, 16, 233–422. [Google Scholar] [CrossRef]
- Woods, C.S. Variation and taxonomic changes in the family Retropinnidae (Salmonoidea). N. Z. J. Mar. Freshw. Res. 1968, 2, 398–425. [Google Scholar] [CrossRef][Green Version]
- Geoghegan, J.L.; Di Giallonardo, F.; Wille, M.; Ortiz-Baez, A.S.; Costa, V.A.; Ghaly, T.; Mifsud, J.C.O.; Turnbull, O.M.H.; Bellwood, D.R.; Williamson, J.E.; et al. Virome composition in marine fish revealed by meta-transcriptomics. Virus Evol. 2021, 7, veab005. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Babaian, A.; Edgar, R.C. Ribovirus classification by a polymerase barcode sequence. bioRxiv 2021. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking virus genomes with host taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Pebesma, E. Simple features for R: Standardized support for spatial vector data. R J. 2018, 10, 439. [Google Scholar] [CrossRef]
- Dunnington, D. ggspatial: Spatial Data Framework for ggplot2. 2021. Available online: https://paleolimbot.github.io/ggspatial/index.html (accessed on 5 December 2021).
- Donovan, P. nzcensr: New Zealands Census Data. 2021. Available online: https://github.com/phildonovan/nzcensr (accessed on 5 December 2021).
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2020. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 5 December 2021).
- Zeileis, A.; Kleiber, C.; Jackman, S. Regression models for count data in R. J. Stat. Softw. 2008, 27, 1–25. [Google Scholar] [CrossRef]
- Jackman, S. pscl: Classes and Methods for R Developed in the Political Science Computational Laboratory. 2020. Available online: https://github.com/atahk/pscl/ (accessed on 5 December 2021).
- Giner, G.; Smyth, G.K. statmod: Probability calculations for the inverse Gaussian distribution. R J. 2016, 8, 339–351. [Google Scholar] [CrossRef]
- Dunn, P.K. Tweedie: Evaluation of Tweedie Exponential Family Models. 2017. Available online: https://cran.r-project.org/web/packages/tweedie/index.html (accessed on 5 December 2021).
- Dunn, P.K.; Smyth, G.K. Evaluation of Tweedie exponential dispersion models using Fourier inversion. Stat. Comput. 2008, 18, 73–86. [Google Scholar] [CrossRef]
- Dunn, P.K.; Smyth, G.K. Series evaluation of Tweedie exponential dispersion models. Stat. Comput. 2005, 15, 267–280. [Google Scholar] [CrossRef]
- Somerfield, P.J.; Clarke, K.R.; Warwick, R.M. Simpson Index. In Encyclopedia of Ecology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 3252–3255. ISBN 9780080914565. [Google Scholar]
- Beck, M.W. ggord: Ordination plots with ggplot2. 2021. Available online: https://fawda123.github.io/ggord/ (accessed on 5 December 2021).
- Kamer, G.; Argos, P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res. 1984, 12, 7269–7282. [Google Scholar] [CrossRef]
- Bosch, A.; Pintó, R.M.; Guix, S. Human Astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048–1074. [Google Scholar] [CrossRef] [PubMed]
- Vike, S.; Nylund, S.; Nylund, A. ISA virus in Chile: Evidence of vertical transmission. Arch. Virol. 2009, 154, 1–8. [Google Scholar] [CrossRef]
- Mekata, T.; Kawato, Y.; Ito, T. Complete genome sequence of Carp edema virus isolated from koi carp. Microbiol. Resour. Announc. 2021, 10, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Garver, K.A.; Leskisenoja, K.; Macrae, R.; Hawley, L.M.; Subramaniam, K.; Waltzek, T.B.; Richard, J.; Josefsson, C.; Tellervo Valtonen, E. An alloherpesvirus infection of european perch Perca fluviatilis in Finland. Dis. Aquat. Organ. 2018, 128, 175–185. [Google Scholar] [CrossRef]
- Ricker, W.E. Computational and interpretation of biological statistics of fish populations. Bull. Fish. Res. Board Can. 1975, 191, 1–382. [Google Scholar]
- Arunkumar, G.A.; Bhavsar, D.; Li, T.; Strohmeier, S.; Chromikova, V.; Amanat, F.; Bunyatov, M.; Wilson, P.C.; Ellebedy, A.H.; Boons, G.J.; et al. Functionality of the putative surface glycoproteins of the Wuhan spiny eel influenza virus. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perry, B.J.; Darestani, M.M.; Ara, M.G.; Hoste, A.; Jandt, J.M.; Dutoit, L.; Holmes, E.C.; Ingram, T.; Geoghegan, J.L. Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition. Viruses 2022, 14, 257. https://doi.org/10.3390/v14020257
Perry BJ, Darestani MM, Ara MG, Hoste A, Jandt JM, Dutoit L, Holmes EC, Ingram T, Geoghegan JL. Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition. Viruses. 2022; 14(2):257. https://doi.org/10.3390/v14020257
Chicago/Turabian StylePerry, Benjamin J., Mitra Mohamadi Darestani, Motia Gulshan Ara, Amélie Hoste, Jennifer M. Jandt, Ludovic Dutoit, Edward C. Holmes, Travis Ingram, and Jemma L. Geoghegan. 2022. "Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition" Viruses 14, no. 2: 257. https://doi.org/10.3390/v14020257
APA StylePerry, B. J., Darestani, M. M., Ara, M. G., Hoste, A., Jandt, J. M., Dutoit, L., Holmes, E. C., Ingram, T., & Geoghegan, J. L. (2022). Viromes of Freshwater Fish with Lacustrine and Diadromous Life Histories Differ in Composition. Viruses, 14(2), 257. https://doi.org/10.3390/v14020257