
The Myotis chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly

Abstract
:1. Introduction
2. Material and Methods
3. Results
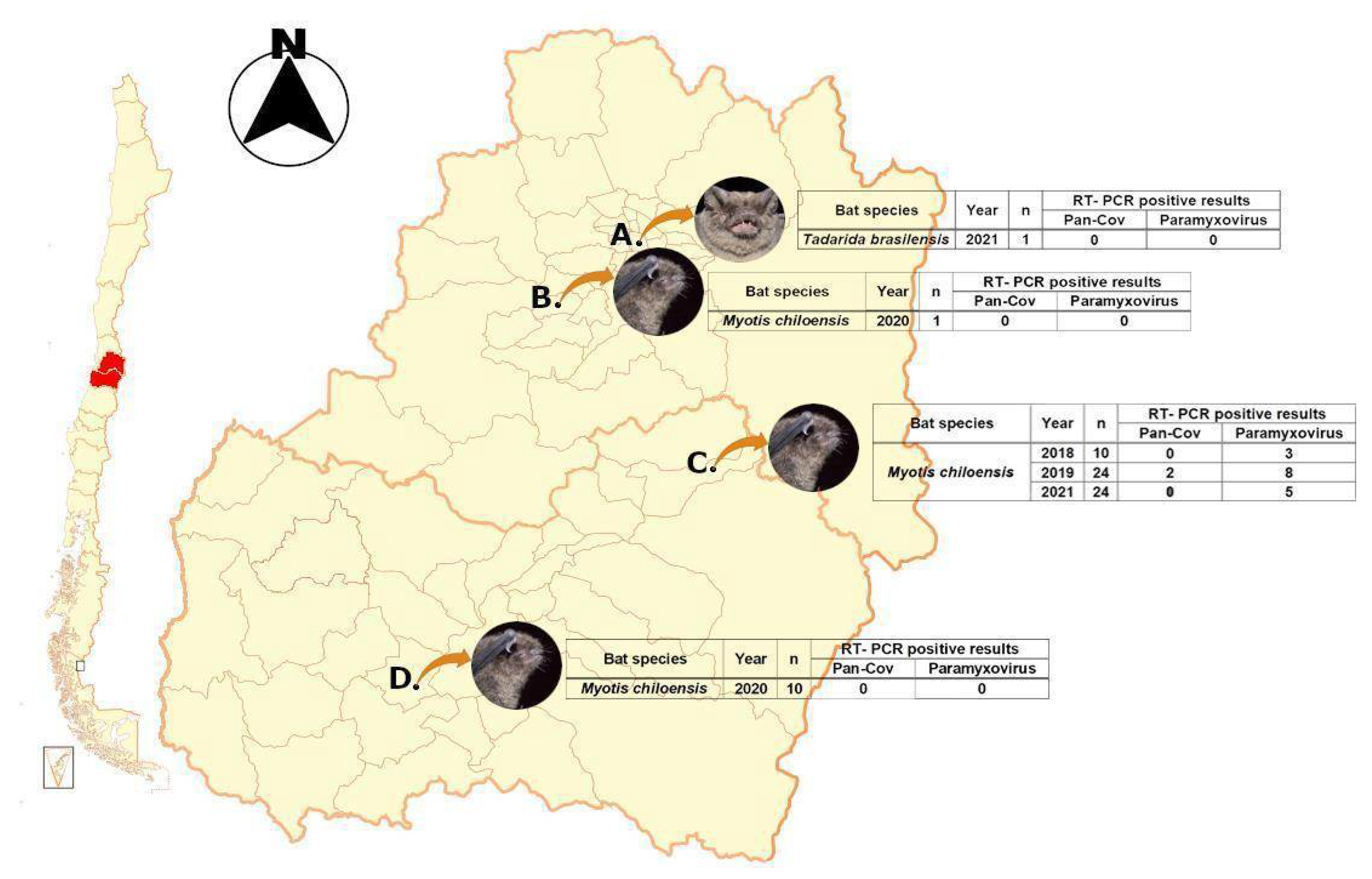
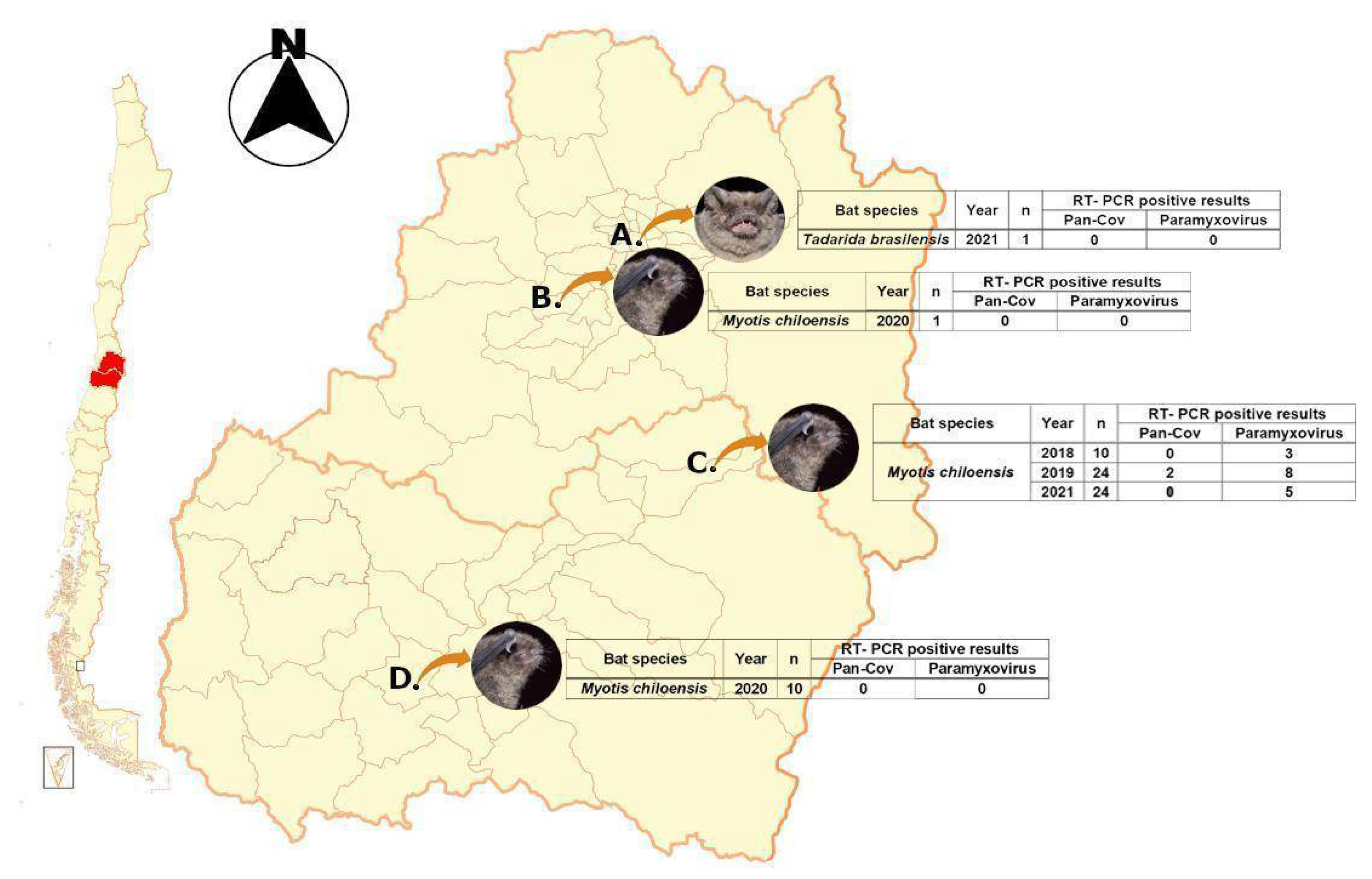
3.1. Collected Samples
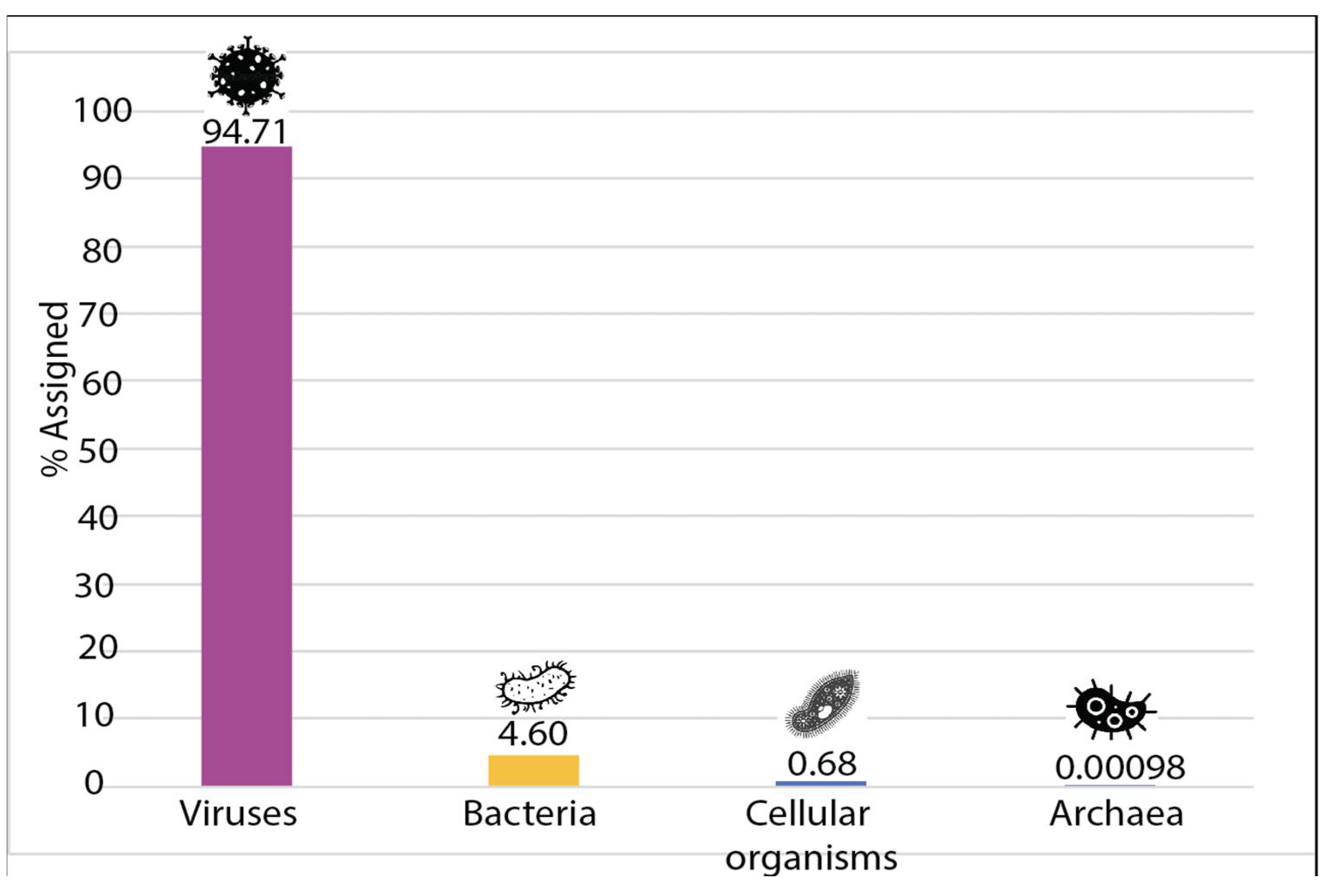
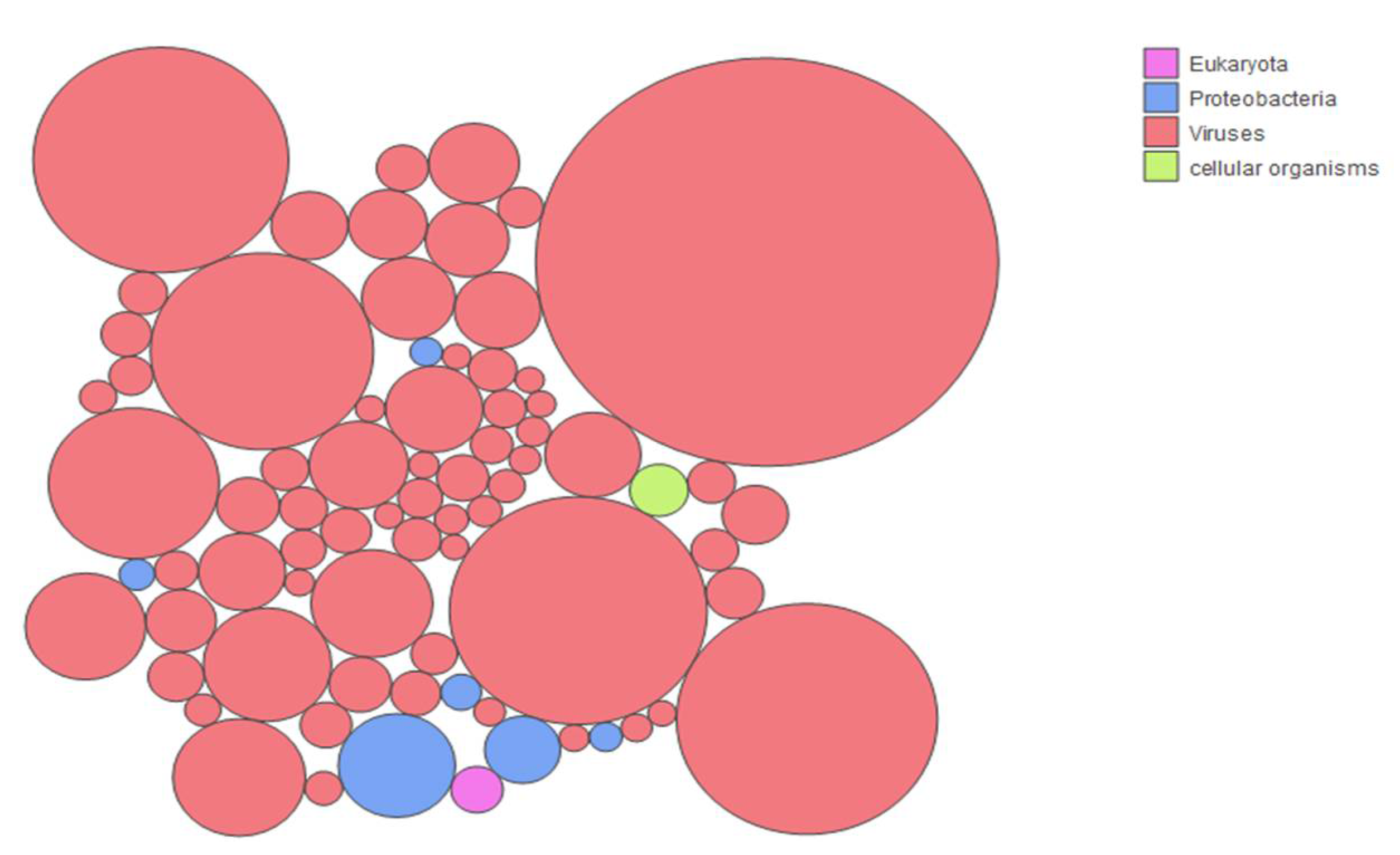
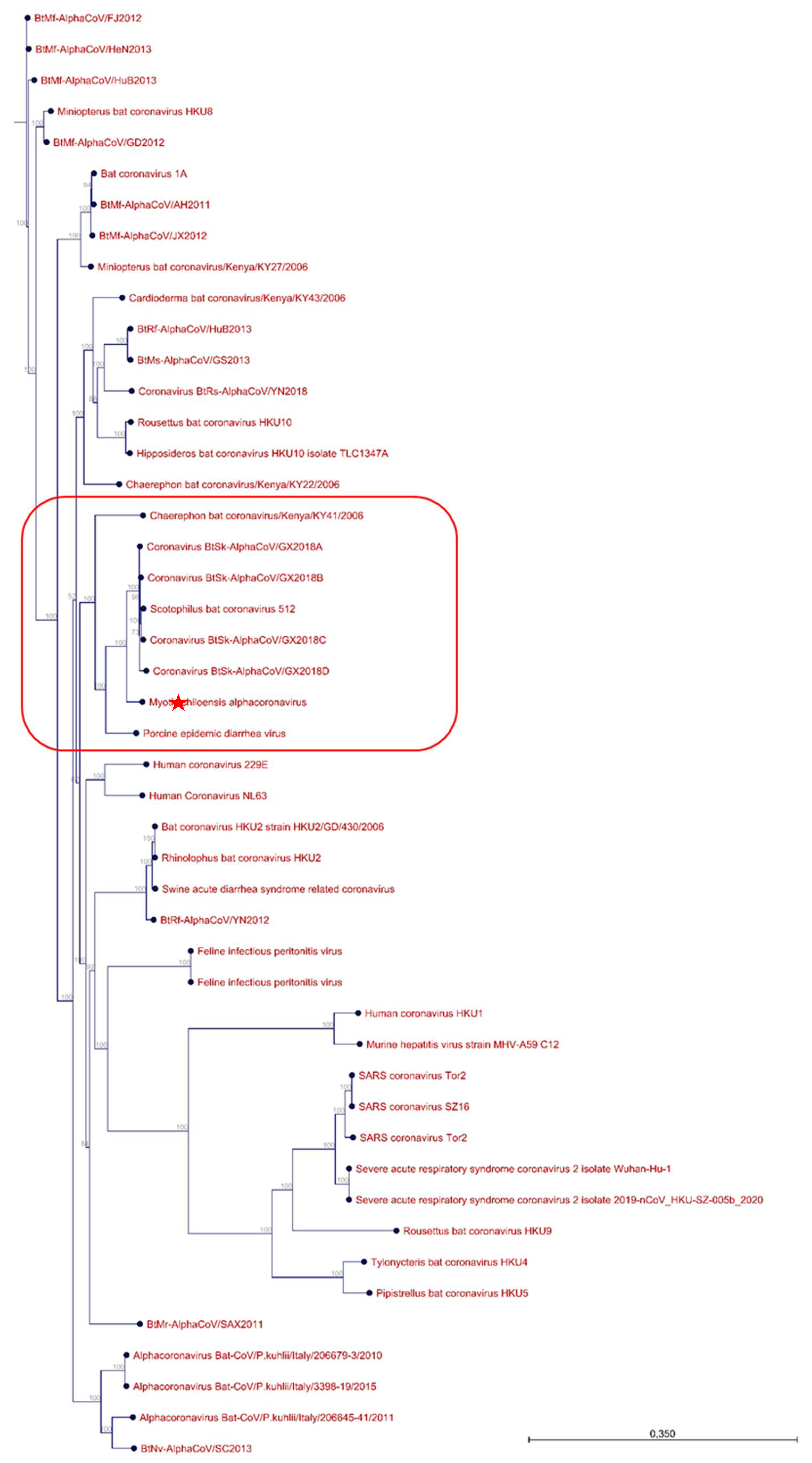
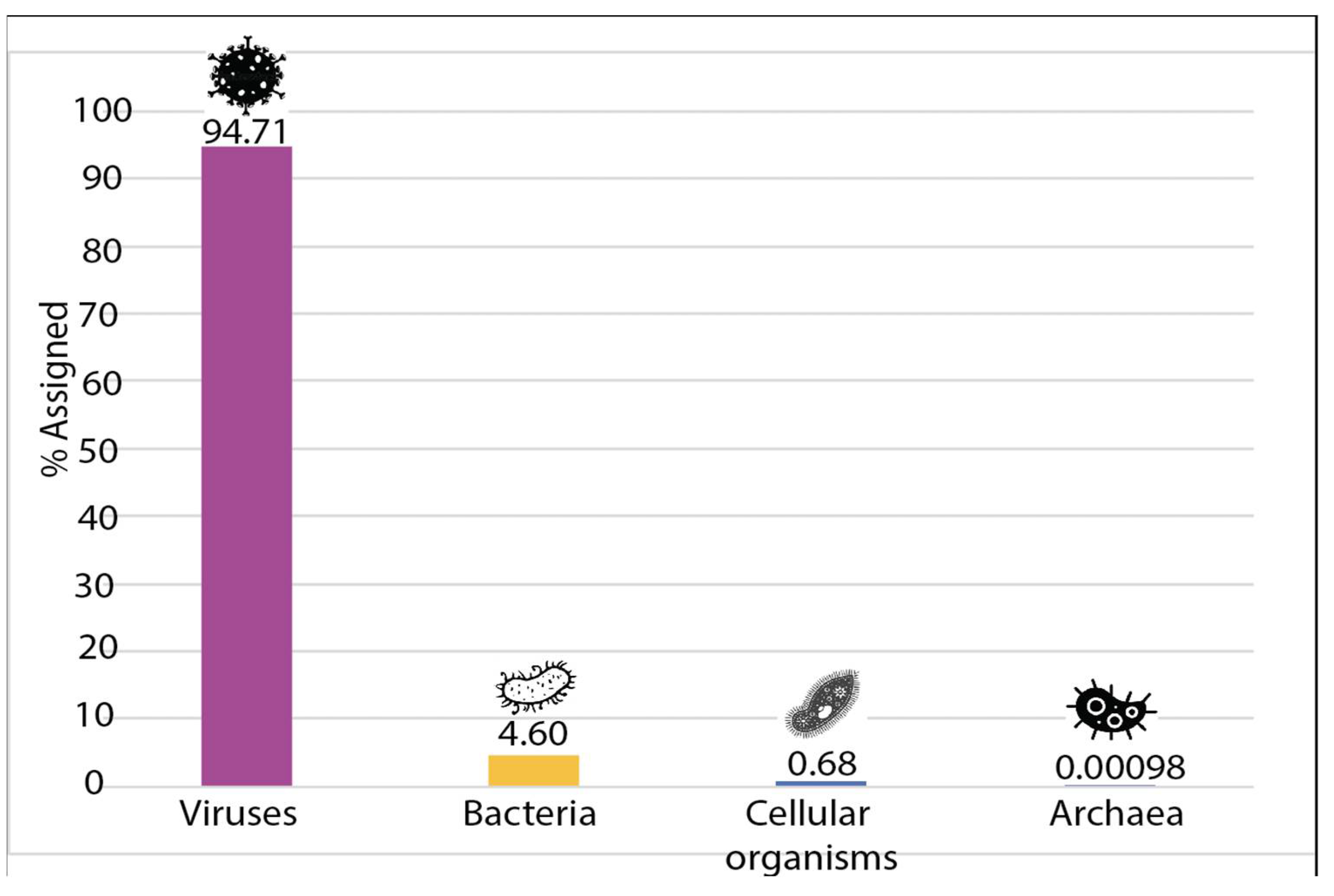
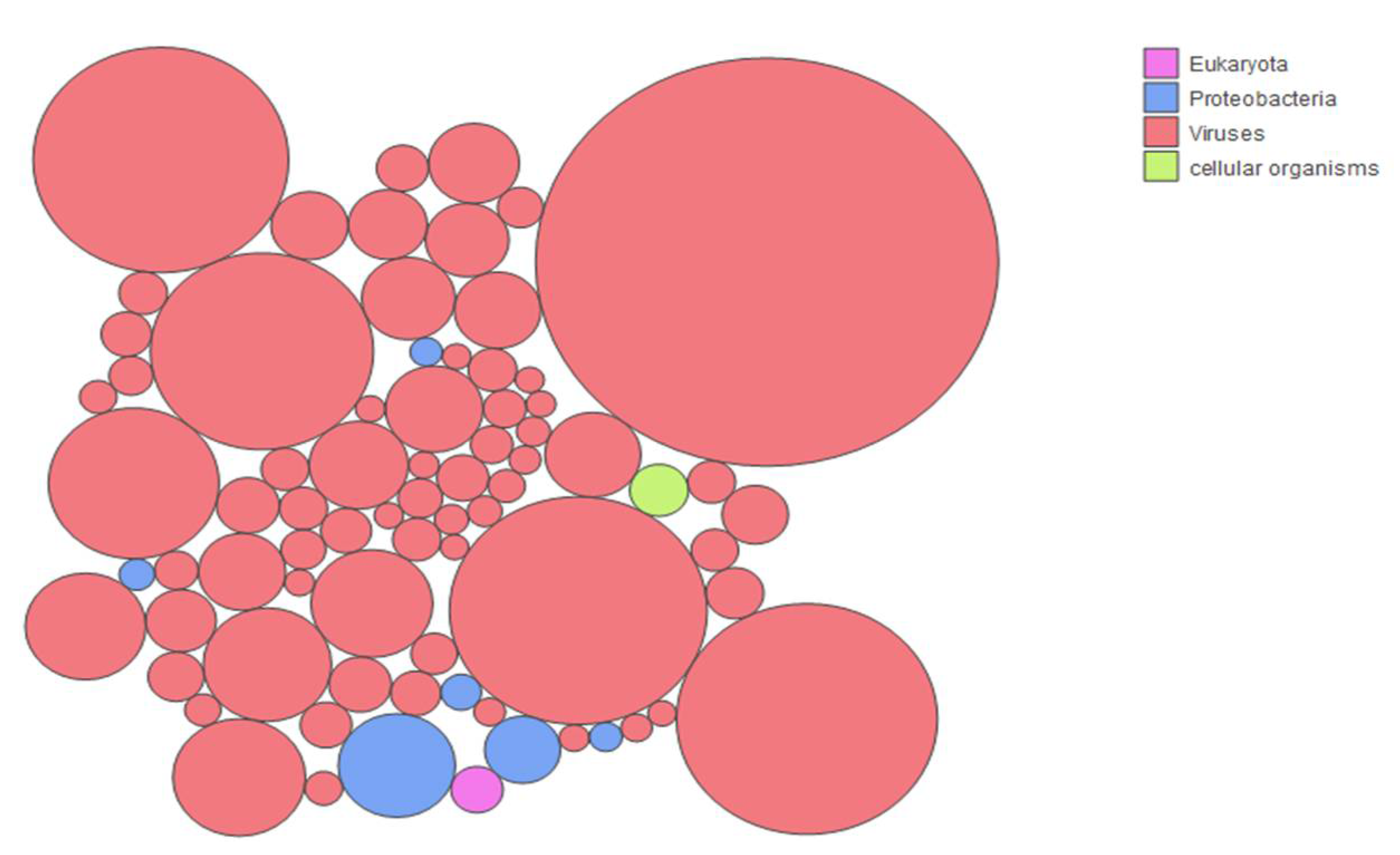
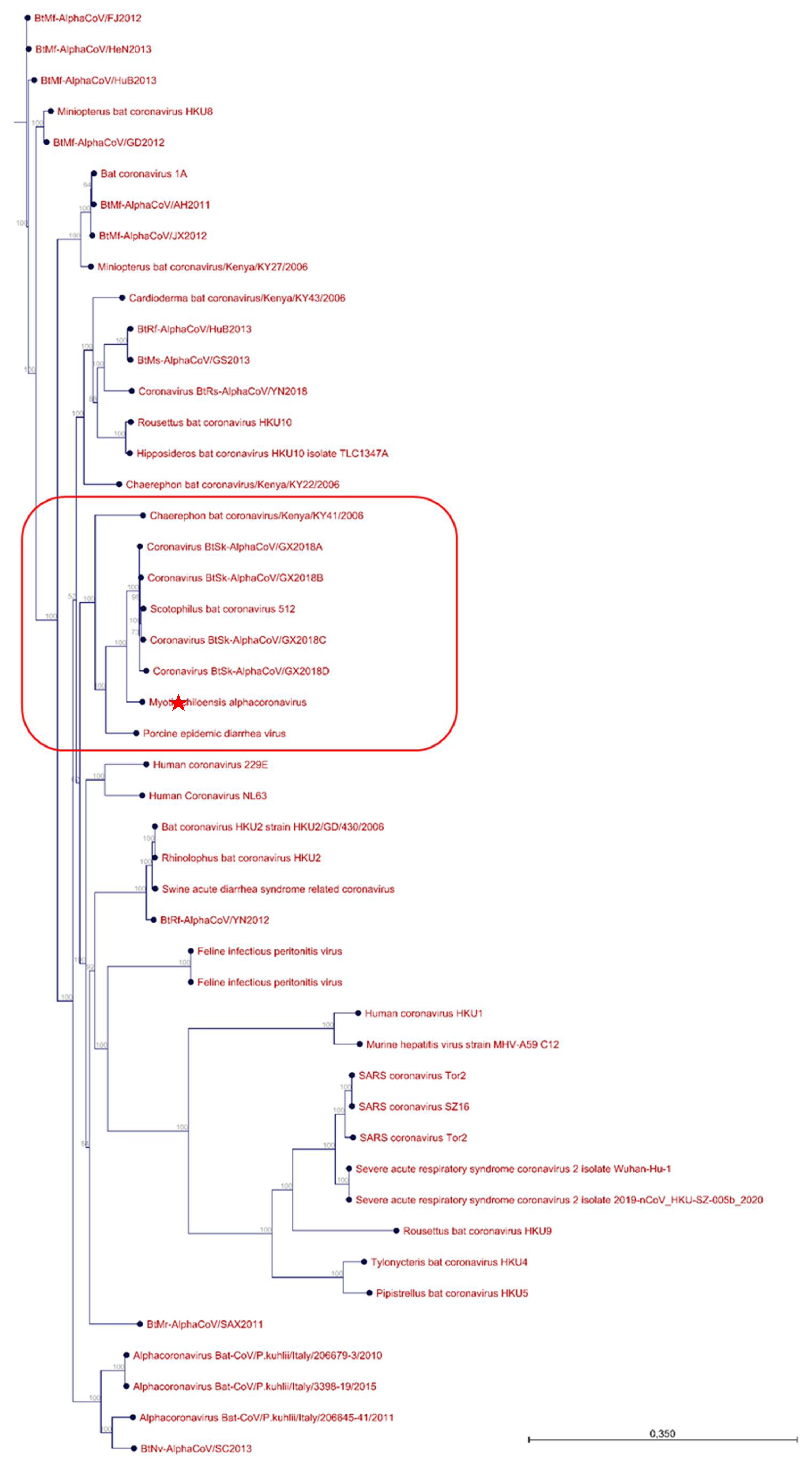
3.2. Sample Enrichment and Virome Elucidation/Alphacoronavirus Genome Assembly and Phylogeny
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, D.E.; Mittermeier, R. Bats. In Handbook of the Mammals of the World; Linx Edicions: Barcelona, Spain, 2019. [Google Scholar]
- Schipper, J.; Chanson, J.S.; Chiozza, F.; Cox, N.A.; Hoffmann, M.; Katariya, V.; Lamoreux, J.; Rodrigues, A.S.; Stuart, S.N.; Temple, H.J.; et al. The Status of the World’s Land. Science 2008, 322, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Kuzmin, I.V.; Bozick, B.; Guagliardo, S.A.; Kunkel, R.; Shak, J.R.; Tong, S.; Rupprecht, C.E. Bats, emerging infectious diseases, and the rabies paradigm revisited. Emerg. Health Threat. J. 2011, 4, 7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, C.C.; Kingston, T. Bats in the anthropocene. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C.C., Kingston, T., Eds.; Springer Open: New York, NY, USA, 2015; pp. 1–9. [Google Scholar]
- Kunz, T.H.; de Torrez, E.B.; Bauer, D.; Lobova, T.; Fleming, T.H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 2011, 1223, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.R. Appraisal of microbial evolution to commensalism and pathogenicity in humans. Clin. Med. Insights Gastroenterol. 2013, 6, CGast-S11858. [Google Scholar] [CrossRef]
- Ossa, G.; Kramer-Schadt, S.; Peel, A.J.; Scharf, A.K.; Voigt, C.C. The Movement Ecology of the Straw-Colored Fruit Bat, Eidolon helvum, in Sub-Saharan Africa Assessed by Stable Isotope Ratios. PLoS ONE 2012, 7, e45729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95, e01713-20. [Google Scholar] [CrossRef]
- Coltart, C.E.M.; Lindsey, B.; Ghinai, I.; Johnson, A.M.; Heymann, D.L. The Ebola outbreak, 2013–2016: Old lessons for new epidemics. Philos. Trans. Royal Soc. B Biol. Sci. 2017, 372, 20160297. [Google Scholar] [CrossRef]
- Benavides, J.A.; Valderrama, W.; Recuenco, S.; Uieda, W.; Suzán, G.; Avila-Flores, R.; Velasco-Villa, A.; Almeida, M.; Andrade, F.A.G.; Molina-Flores, B.; et al. Defining New Pathways to Manage the Ongoing Emergence of Bat Rabies in Latin America. Viruses 2020, 12, 1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef]
- Shi, J.; Sun, J.; Hu, N.; Hu, Y. Phylogenetic and genetic analyses of the emerging Nipah virus from bats to humans. Infect. Genet. Evol. 2020, 85, 104442. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossa, G.; Rodriguez-San Pedro, A. Myotis chiloensis. Mamm. Species 2015, 47, 51–56. [Google Scholar] [CrossRef]
- Wilkins, K.T. Tadarida brasiliensis. Mamm. Species 1989, 331, 1–10. [Google Scholar] [CrossRef]
- Hayman, D.T.S.; Bowen, R.A.; Cryan, P.M.; McCracken, G.F.; O’shea, T.J.; Peel, A.J.; Gilbert, A.; Webb, C.T.; Wood, J.L.N. Ecology of Zoonotic Infectious Diseases in Bats: Current Knowledge and Future Directions. Zoonoses Public Health 2013, 60, 2–21. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef] [PubMed]
- Barquez, R.M.; Diaz, M.M. Myotis chiloensis. In IUCN Red List of Threatened Species; IUCN: Cambridge, UK, 2008. [Google Scholar]
- Mann, G. Los pequeños mamiferos de Chile. Gayana Concepción 1978, 40, 1–342. [Google Scholar]
- Ossa, G.; Lilley, T.M.; Waag, A.G.; Meierhofer, M.B.; Johnson, J.S. Roosting ecology of the southernmost bats, Myotis chiloensis and Histiotus magellanicus, in southern Tierra del Fuego, Chile. Austral. Ecol. 2020, 45, 1169–1178. [Google Scholar]
- Mares, M.A.; Bárquez, R.M.; Braun, J.K.; Barquez, R.M.; Braun, J.K. Distribution and ecology of some Argentine bats (Mammalia). Ann. Carnegie Mus. 1995, 64, 219–237. [Google Scholar]
- Hoyt, J.R.; Kilpatrick, A.M.; Langwig, K.E. Ecology and impacts of white-nose syndrome on bats. Nat. Rev. Microbiol. 2021, 19, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Vijgen, L.; Moës, E.; Keyaerts, E.; Li, S.; van Ranst, M. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol. Biol. 2008, 454, 3–12. [Google Scholar] [CrossRef] [Green Version]
- van Boheemen, S.; Bestebroer, T.M.; Verhagen, J.H.; Osterhaus, A.D.; Pas, S.D.; Herfst, S.; Fouchier, R.A. A family-wide RT-PCR assay for detection of paramyxoviruses and application to a large-scale surveillance study. PLoS ONE 2012, 7, e34961. [Google Scholar] [CrossRef] [Green Version]
- Calgua, B.; Mengewein, A.; Grunert, A.; Bofill-Mas, S.; Clemente-Casares, P.; Hundesa, A.; Wyn-Jones, A.P.; López-Pila, J.M.; Girones, R. Development and application of a one-step low cost procedure to concentrate viruses from seawater samples. J. Virol. Methods 2008, 153, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Paskey, A.C.; Ng, J.H.; Rice, G.K.; Chia, W.N.; Philipson, C.W.; Foo, R.J.; Cer, R.Z.; Long, K.A.; Lueder, M.R.; Frey, K.G.; et al. The temporal RNA virome patterns of a lesser dawn bat (Eonycteris spelaea) colony revealed by deep sequencing. Virus Evol. 2020, 6, veaa017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmier, A.; Tirera, S.; de Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PLoS ONE 2017, 12, e0186943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.; Wang, L.-F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Qin, P.; Wang, B.; Liu, Y.; Xu, G.H.; Peng, L.; Zhou, J.; Zhu, S.J.; Huang, Y.W. Broad Cross-Species Infection of Cultured Cells by Bat HKU2-Related Swine Acute Diarrhea Syndrome Coronavirus and Identification of Its Replication in Murine Dendritic Cells In Vivo Highlight Its Potential for Diverse Interspecies Transmission. J. Virol. 2019, 93, e01448-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simas, P.V.M.; de Souza Barnabé, A.C.; Durães-Carvalho, R.; de Lima Neto, D.F.; Caserta, L.C.; Artacho, L.; Jacomassa, F.A.F.; Martini, M.C.; Dos Santos, M.M.A.B.; Felippe, P.A.N.; et al. Bat coronavirus in Brazil related to appalachian ridge and porcine epidemic diarrhea viruses. Emerg. Infect. Dis. 2015, 21, 729–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Li, J.; Zhou, Q.; Xu, Z.; Chen, L.; Zhang, Y.; Xue, C.; Wen, Z.; Cao, Y. A New Bat-HKU2-like Coronavirus in Swine, China, 2017. Emerg. Infect. Dis. 2017, 23, 1607–1609. [Google Scholar] [CrossRef] [Green Version]
- Steinel, A.; Parrish, C.R.; Bloom, M.E.; Truyen, U. Parvovirus infections in wild carnivores. J. Wildl. Dis. 2001, 37, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Vongphayloth, K.; Salazar, E.B.; Munier, S.; Bonomi, M.; Régnault, B.; Douangboubpha, B.; Karami, Y.; Chretien, D.; Sanamxay, D.; et al. Coronaviruses with a SARS-CoV-2-like receptor- binding domain allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. Res. Sq. 2021; in press. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publication | % Viral Sequences | Enrichment Method |
|---|---|---|
| Wu et al. [29] | 0.8 | Centrifugation, filtration, nuclease treatment, and QIAmp MinElute Virus Spin Kit |
| Salmier et al. [31] | 0.3 | Centrifugation, filtration, nuclease treatment, and NucliSENS easyMAG® bio-robot |
| Wu et al. [28] | 1.2 | Centrifugation, filtration, nuclease treatment, and QIAamp viral RNA minikit |
| Paskey et al. [30] | 0.226 | QIAGEN RNeasy Kit with on-column DNase digestion |
| Aguilar et al., this study (2022) | 2.2 | Centrifugation, filtration, nuclease treatment, QIAamp viral RNA minikitand Magelia with Ribozero Plus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar Pierlé, S.; Zamora, G.; Ossa, G.; Gaggero, A.; Barriga, G.P. The Myotis chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly. Viruses 2022, 14, 202. https://doi.org/10.3390/v14020202
Aguilar Pierlé S, Zamora G, Ossa G, Gaggero A, Barriga GP. The Myotis chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly. Viruses. 2022; 14(2):202. https://doi.org/10.3390/v14020202
Chicago/Turabian StyleAguilar Pierlé, Sebastian, Gabriel Zamora, Gonzalo Ossa, Aldo Gaggero, and Gonzalo P. Barriga. 2022. "The Myotis chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly" Viruses 14, no. 2: 202. https://doi.org/10.3390/v14020202
APA StyleAguilar Pierlé, S., Zamora, G., Ossa, G., Gaggero, A., & Barriga, G. P. (2022). The Myotis chiloensis Guano Virome: Viral Nucleic Acid Enrichments for High-Resolution Virome Elucidation and Full Alphacoronavirus Genome Assembly. Viruses, 14(2), 202. https://doi.org/10.3390/v14020202