1. Introduction
The Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is an enveloped, positive sense, single-stranded RNA virus. It belongs to the
Betacoronaviriae genus, and it was first detected in China in December 2019. As it is generally known, RNA viruses show relatively high mutational rates [
1,
2,
3,
4], and despite the proof reading-repair 3′ to 5′exonuclease mechanism present in some coronaviruses [
5,
6,
7,
8], SARS-CoV-2 acquires mutations in each replication cycle, giving rise to new lineages. According to the definition given by Centers for Disease Control and Prevention (CDC) in Atlanta (USA), a new lineage is considered as a group of viruses closely related sharing a common ancestor (
https://www.ecdc.europa.eu/en/COVID-19/variants-concern, accessed on 10 October 2022). All the lineages described to date (October 2022) may cause COVID-19, and many of them have been referred to as variants of concern (VOC), variant of interest (VOI), or variant being monitored (VBM) [
9] due to the acquisition of mutations associated. For example, changes on the receptor binding domain are expected to affect functional properties, altering infectivity capacity, disease severity, and/or interactions with host immunity leading to immune escape [
10,
11,
12,
13]. Since the beginning of the pandemic, and with the first reports of the SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank accession no: MN908947) [
14], hundreds of new variants have been described in different geographic regions [
10,
11,
15].
Omicron (lineages B.1.1.529 and BA.x) and Delta (lineages B.1.617.2 and AY.x) were the only two variants considered as VOC at the time of writing this article, while variants Alpha (lineages B.1.1.7 and Q.x), Beta (Lineages B.1.351.1, B.1.351.2 and B.1.351.3), Gamma (Lineage P.1 and P.1.x), Epsilon (lineages B.1.427 and B.1.429), Eta (lineage B.1.525), Iota (lineage B.1.526), Kappa (lineage B.1.617.1), Zeta (lineage P.2), Mu (lineages B.1.621 and B.1.621.1), Lambda (C.37 and C.37.1), and lineage B.1.617.3 are now considered as variants being monitored (VBM) because their expansion has been recently diminished and controlled to a great extent. SARS-CoV-2 genome evolution has been analysed since 2020, and several sets of mutations have emerged in the context of variants that impact virus characteristics, including transmissibility and antigenicity [
10,
13,
16]. Most mutations determining VOC or VOI are affecting the spike protein (encoded by the S gene), a primary antigen of SARS-CoV-2 [
17] related to infective capacity and immunological responses [
13,
18,
19].
Lineage identification has been addressed in several methods, mainly strategies based on PCR targeting single or very few nucleotide mutations [
20]. However, single-nucleotide mutations (SNPs) are not always SARS-CoV-2 variant-specific, and a large number of SNPs need to be typed for reliable identification of each lineage. Whole-genome sequencing (WGS) has been considered as the gold-standard method to determine SARS-CoV-2 variants; nevertheless, this technology is complex and expensive, not affordable for most laboratories. Since the beginning of 2021, an action protocol named Spike Gene Target Failure (SGTF) was extended to identify the Alpha variant (B.1.1.7). According to this protocol, a negative result obtained from the PCR targeting a region of S-gene (recommended to be tested simultaneously with SARS-CoV-2 diagnostic PCR) should be taken as symptom of presumptive Alpha variant and the sample should be investigated by WGS. By using criteria along these lines, the diversity described may be bypassed to sequences containing the mutation responsible for the S-gene PCR-negative result, ignoring other sequences probably belonging to Alpha variant.
In the present article, we report a fast, simple, and affordable strategy to identify the most relevant SARS-CoV-2 lineages described so far. The method is based on the Sanger-sequencing of a 921 bp S-gene fragment comprising 30 informative variable sites rather than these based on the complete or almost complete sequencing of the S-gene or whole-genome sequencing, as previously reported [
21,
22,
23,
24]. The strategy proposed in the present study can be performed with only a single PCR and a simple analysis, constituting a cost-effective methodology for most diagnostic laboratories. The reagents and protocol have been made available world-wide by Genetic PCR Solutions™ (GPS™, Alicante, Spain), also providing a simple graphical user interface (GUI) and a reference database including reference genomes from all variants concerned.
2. Materials and Methods
2.1. Determination of Informative Nucleotide Positions from Genome Analysis
Ten complete genomes of each variant, namely VOC Omicron and Delta, VBM Alpha, Beta, Gamma, Epsilon, Eta, Iota, Kappa, Lambda, and Mu, and lineages B.1.617.3, A.23.1, C.16, B.1.258, and B.1.1.207 were downloaded from the GISAID database and evaluated to determine the presence of mutations. Different genomes from separate laboratories at diverse world regions, excluding redundant data, not complete data, and low-coverage genomes, were selected to maximize diversity for each variant. Individual genes (ORF1ab, S, 3a, E, M, Orf6, Orf7a, Orf7b, Orf8, N, and Orf10) were identified on the alignment by comparison with the reference genome (NC_045512.2) retrieved from the National Center for Biotechnology information (NCBI) website databases (Bethesda, MD, USA). Multiple sequence alignment was performed with ClustalW algorithm, and the resulting alignments were analysed for identification of variant-specific nucleotides by using the Phylogenetic Analysis option on the Mega 11.0.10 software [
25]. Only single-nucleotide mutations involving amino acid changes were considered (
Table A1). Furthermore, selected mutations were thoroughly evaluated for each variant using all available sequences at GISAID database deposited until July 2022 (12,349,470 sequences). In this step of the analysis, not only individual mutations were evaluated but also their combinations: the aim was to determinate variant-specific signature sequences. Subsequently, the emerging Omicron sub-lineages were analysed to evaluate the discrimination capacity of the fragment.
2.2. Preparative End-Point Reverse Transcriptase-PCR
Specific sets of primers flanking a selected 921 bp fragment of the S-gene and all reagents used to perform the reverse-transcriptase end-point PCR (RT-epPCR) were supplied in the SARS-CoV- 2 seq-RT-epPCR kit (GPS™, Alicante, Spain) [
26,
27], following manufacturer’s instructions. RT-epPCR reactions were initially carried out using 10
5 copies/µL of SARS-CoV-2 synthetic RNA Control 2 Wuhan-Hu-1 (GenBank ID MN908947.3) from Twist Bioscience (South San Francisco, USA). All the PCRs were performed with a final volume of 25 µL, containing 6 µL of GPS™-mix-RT (5×), 1 µL of primer-probe mix, 16 µL of H
2O, and 2 µL of sample. The one-step amplification regime included 10 min of retrotranscription at 48 °C, an activation step of 2 min at 95 °C, and 35 cycles composed by a denaturation step at 95 °C for 5 s, 30 s at 54 °C for annealing, and 90 s at 72 °C for the extension. A final elongation step was added, lasting 5 min at 72 °C. Samples were analysed in an agarose gel (agarose D1 LOW EEO, Pronadisa) using 4 µL of PCR product and 2 µL of EZVision (VWR Life science) running for 30 min with 115 V.
2.3. Partial S-Gene Sequencing and Phylogenetic Analysis
All the samples with a band visible to the naked eye on the agarose gel were purified using GPSpin PCR Kit (GPS™, Alicante, Spain) and eluted with 30 µL of DNase/RNase free water. The amount (ng/µL) of DNA was determined studying 2 µL of the sample in the spectrophotometer DS-11 (Denovix, Wilmington, DE, USA). Between 10–30 ng/µL of each purified PCR product and sequencing primers at 5 µM were mixed with a final volume of 10 µL and submitted to GATC (Eurofins Genomics, Coralville, IA, USA) services for Sanger-based sequencing. Resulting sequences were analysed by comparison with a S-gene sequence reference database. Five S-gene 921 bp fragment sequences of each variant were included into this reference database, corresponding with the most diverse genomes for each variant (
Table A2) harvested from the GISAID public database. Alignment and phylogenetic analysis were performed by using the Mega 11.0.10 software [
25] with the neighbour-joining method [
28] and bootstrap values for 1000 replicates. Alongside this, phylogenetic analysis of obtained sequences and sequences retrieved from the reference database were achieved by using a newly developed program, which is available with a simple graphical user interface (GPS™, Alicante, Spain) following the designer’s instructions.
2.4. Analytical Validation
Preliminary experimental assays were carried out with 105 copies/µL of SARS-CoV-2 synthetic RNA Control 2 Wuhan-Hu-1 (GenBank ID MN908947.3) from Twist Bioscience (South San Francisco, CA, USA). Furthermore, analytical validation included the testing of RNA extracts from 129 clinical SARS-CoV-2-positive samples provided by two external clinical laboratories. It was confirmed by using the GPS™ COVID-19 dtec-RT-qPCR kit (Alicante, Spain).
3. Results
3.1. In Silico Specificity of the S-Gene 921 BP-Fragment
The sequence analysis of ten complete genomes of each variant, i.e., Omicron, Delta, Alpha, Beta, Gamma, Epsilon, Eta, Iota, Kappa, Lambda, and Mu, and lineages B.1.617.3, A.23.1, C.16, B.1.258, and B.1.1.207 (GISAID) confirmed that S-gene concentrates a considerable number of mutations compared to other parts of the genome. The fragment of 921 bp, ranging from positions 22,784–23,705 of the reference genome Wuhan-Hu-1 (NCBI ref.: NC_045512.2) comprised 30 variable nucleotide positions whose composition was considerably specific for the variants under analysis. All 30 mutations of each variant were evaluated against 12,250,005 genomes (GISAID, July 2022); afterwards, the frequencies were calculated (shown in
Appendix A). Finally, the combination of these mutations yielded a high level of in silico inclusiveness (proportion of genome sequences of a determined variant containing the corresponding specific mutation pattern) and exclusiveness (% of sequences of a variant containing a specific mutation pattern compared to all SARS-CoV-2 genomes containing the specified pattern) for all the variants subjected to this search (
Table 1). Therefore, the sequence of this 921-fragment was considered informative to determine SARS-CoV-2 variants and was subjected to phylogenetic analysis. The resulting neighbour-joining phylogenetic tree obtained using a simplified database containing five S-gene 921 bp fragment sequences for each variant (
Table A2) showed clearly separated clusters for each SARS-CoV-2 variant (
Figure 1). With this methodology, Omicron sub-lineages BA.4 and BA.5 as well as XE and BA.2 could not be differentiated from each other although all of them fell within the cluster corresponding to the Omicron variant.
3.2. In Vitro PCR-Amplification, Sequencing, and Phylogenetic Analysis of the Assay
Optimization of the RT-epPCR protocol was performed by selecting the most suitable set of primers, which is supplied in the SARS-CoV- 2 seq-RT-epPCR kit (GPS™, Alicante, Spain) and 10
5 copies/µL of reference SARS-CoV-2 synthetic RNA Control 2 Wuhan-Hu-1 (GenBank ID MN908947.3). The optimized RT-epPCR protocol was applied to purified RNA from clinical samples previously determined as SARS-CoV-2-positive and confirmed in our laboratory by real-time PCR. Amplified PCR products were analysed by agarose electrophoresis, and the expected bands corresponding to the 921 bp fragment size were visualized in an UV-transilluminator.
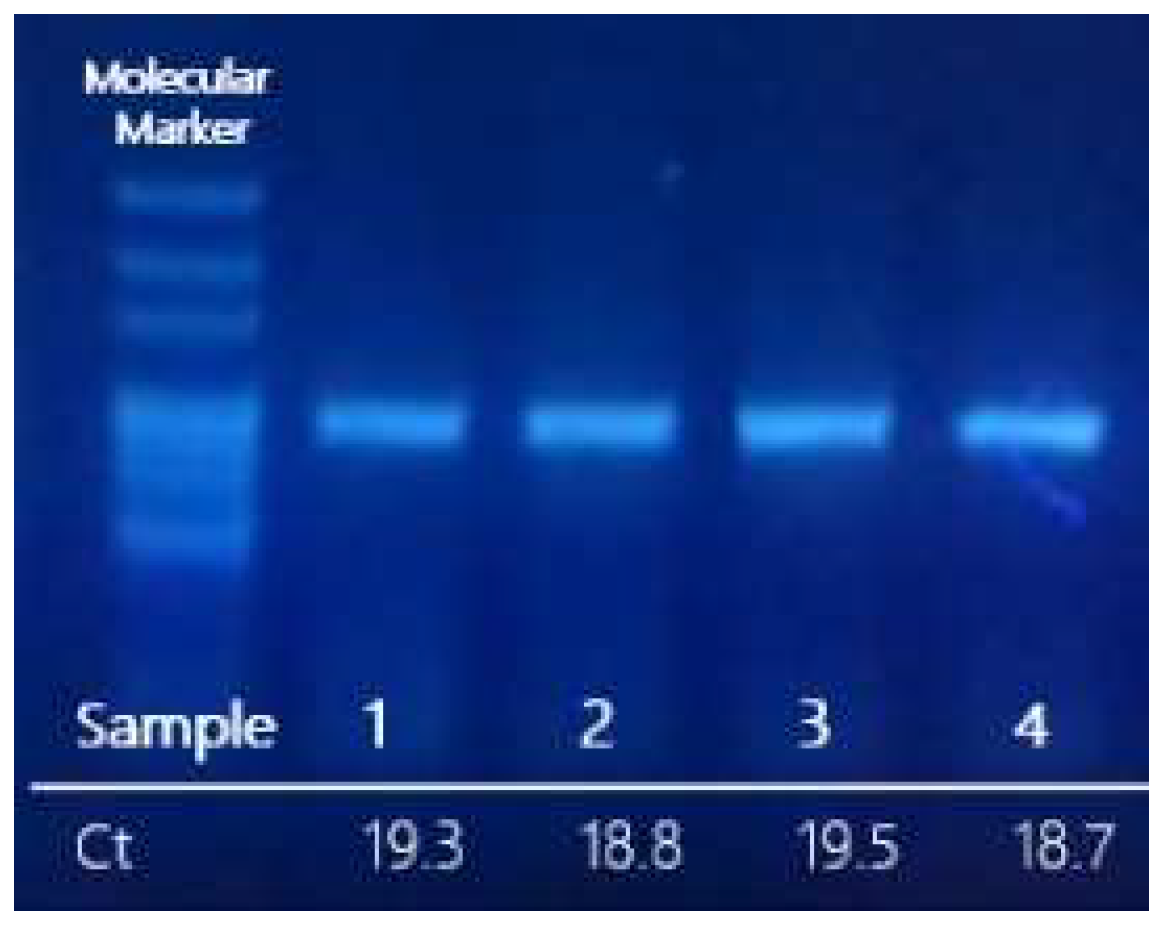
Figure 2 shows the results obtained from clinical samples (2021/SARS-CoV-2/GPS001, 2021/SARS-CoV-2/GPS002, 2022/SARS-CoV-2/GPS003, and 2022/SARS-CoV-2/GPS004). Samples with low RNA concentration (Ct > 29 in qPCR, below 1000 copies) may not yield enough amplicons for sequencing.
Purified products of the RT-epPCR were subjected to sequencing, and the resulting sequence data were processed as previously described using our reference database to obtain a phylogenetic tree for variant determination (
Figure 1). In the resulting tree, the sequences from analysed samples clustered with their corresponding variant as shown in
Figure 2: sample 2021/SARS-CoV-2/GPS001 clustered with the B.1.1.7 lineage (variant Alpha); sample 2021/SARS-CoV-2/GPS002 with B.1.617.2 lineage (variant Delta); and samples 2022/SARS-CoV-2/GPS003 and 2022/SARS-CoV-2/GPS004 with cBA.4/BA.5/B.1.529 lineages (variant Omicron). The same result was obtained when using a simple graphical user interface recently developed (GPS™, Alicante, Spain) that automatically assembles, aligns, and generates a phylogenetic tree.
3.3. Analytical Validation of the Method
The developed method for SARS-CoV-2 variant identification was applied to RNA extracted from a COVID-19-positive clinical samples received by our laboratory (
Table 2) and corroborated in our laboratory by real-time PCR (not shown). Samples collected during early period April–June 2021 showed a clear tendency to be identified as Alpha variant. This trend changed throughout July, with a clear twist towards the Delta variant, which reached its pinnacle during August 2021. A few additional samples collected in 2022 were identified as Omicron variant. Finally, five samples could not be assigned to any variant by the assay, representing 3.9% of samples analysed (5/129), as they may belong to other variants not included in our database.
4. Discussion
A considerable number of SARS-CoV-2 variants have emerged during the last two years, but only a few acquired mutations affect the pathogenic characteristics of the virus. Among all of them, the Omicron (lineages B.1.1.529 and BA.x), Delta (lineages B.1.617.2 and AY.x), and Alpha (lineages B.1.1.7 and Q.x) have spread worldwide to such point they represent ca. 90% of all genomes deposited into GISAID. Current lineage identification has been addressed by two main strategies: qPCR-based targeting of single-nucleotide mutations or whole-genome sequencing. In our laboratory, a review of some single-nucleotide mutations of SARS-CoV-2 (∆69–70, N501Y, and ∆144) revealed that they were not always variant-specific as expected. For instance, mutation ∆69–70 has been largely considered exclusive for Alpha variant; however, by February 2021, only 82% of known SARS-CoV-2 genomes containing mutation ∆69–70 were Alpha, while the remaining 18% corresponded to several other variants (data not shown). Afterwards, the number of Alpha genomes increased exponentially, which contributed to artificially increase the degree of in silico specificity of the mutation. Consequently, a proper lineage identification required the simultaneous analysis of multiple single-nucleotide mutations, making the qPCR approach more complex. It is generally accepted that whole-genome sequencing (WGS) is considered as the gold-standard method to determine SARS-CoV-2 variants. Nevertheless, this technology is complex and expensive, not affordable for most laboratories; therefore, an artificial criterion to identify the Alpha variant (B.1.1.7) was stablished to select the samples of interest for WGS in well-equipped reference laboratories. The protocol was named Spike Gene Target Failure (SGTF) and was based in a qPCR targeting ∆69–70 of the S-gene, assayed simultaneously with SARS-CoV-2 diagnostic PCR. According to this protocol, a negative result in the qPCR targeting ∆69–70 should be considered as presumptive Alpha variant, and the sample must be submitted to WGS analysis. Unfortunately, this criterion generated a biased enrichment of ∆69–70 Alpha genome sequences on databases and could have masked other populations of minority lineages.
This new strategy for SARS-CoV-2 variant identification based on partial S-gene sequencing may solve the issues derived from the information provided by single-nucleotide mutation PCR, also avoiding the complexity/expenses of WGS. The simplicity makes the method described here a candidate tool to be implemented in laboratories of control, without a need for outstanding skills and investments. Results revealed that the presence of a specific combination of mutations in the selected short fragment allowed the identification of several relevant variants, including VOC Delta and Omicron and VBM Alpha, Beta, Gamma, Eta, and Lambda, with high levels of inclusivity and exclusivity (
Table 1).
Although all sub-lineages of Omicron were grouped in a single cluster including the first lineage B.1.1.529, some of them showed a very close phylogenetic relationship and could not be differentiated form each other, for instance, BA.4 and BA.5 as well as XE and BA.2. The identification results retrieved from the RNA extracts of 129 SARS-CoV-2-positive clinical samples revealed a good correlation with the predominant variant circulating during the time of sampling, as the collection was performed through several COVID-19 waves. The Ministry of Health of Spain has reported predominance of the Alpha variant since the beginning of 2021, an increase in the Delta variant from July 2021 to December 2021, and the emergence of the Omicron variant during January 2022.
Controlling the spread of emergent variants could be crucial in the fight against SARS-CoV-2. The assay developed and described in the present article, available from GPS™ (Alicante, Spain), allows a quick and affordable identification of the main variants catalogued as VOC/VOI by competent entities. This tool could provide resources to laboratories that do not have the technological capacity to undertake WGS, without the need to restrict the number of samples through previous criteria, hence being a massive contribution.
5. Conclusions
The appearance of new viral variants, such as those of SARS-CoV-2, is a natural viral process, which can lead to diagnosis failures and treatment modifications. The possibility to easily track lineages would shed light into the spread of the SARS-CoV-2 disease but also in the monitoring of VOC, VOI, and VBM through their expansion, alerting authorities and health care workers of new possible outbreaks. This developed strategy for SARS-CoV-2 variant identification based on partial S-gene sequencing could simplify virus monitoring since it is a fast and reliable strategy to evaluate every SARS-CoV-2-positive sample detected through a manageable and affordable analysis.
Author Contributions
Methodology, A.M.-M., A.G.-S., A.N. and L.P.; Investigation, A.M.-M., A.G.-S., A.N. and L.P.; Data curation, A.M.-M., A.G.-S., A.N. and L.P.; Writing—original draft, A.M.-M., A.G.-S., A.N. and L.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto Valenciano de Competitividad Empresarial (IVACE); program PIDI-CV, grant number IMIDTA/2021/75.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data have been included in the text.
Acknowledgments
This study was possible thanks to GPS™ financial support and to Paloma Romero-Agulló for the review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Table A1.
The table shows the inclusivity for the combination of all mutations in each variant, according to data obtained from GISAID in October 2022. In green, inclusivity percentages above 80% are highlighted.
Table A1.
The table shows the inclusivity for the combination of all mutations in each variant, according to data obtained from GISAID in October 2022. In green, inclusivity percentages above 80% are highlighted.
| | Omicron | Delta | Alpha | Beta | Gamma | Epsilon | Eta | Iota | Zeta | Kappa | Lambda | Mu | - | - | - | - | - |
|---|
| Mutation | B.1.1.529 | BA.4 | BA.5 | Omicron XE + BA.2 | B.1.617.2 + AY.x | B.1.1.7 + Q.x | B.1.351 + B.1.351.2 + B.1.351.3 | P.1 + P.1.x | B.1.427 + B.1.429 | B.1.525 | B.1.526 | P.2 | B.1.617.1 | C.37 + C.37.1 | B.1.621 + B.1.621.1 | B.1.617.3 | A.23.1 | C.16 | B.1.258 | B.1.1.207 |
|---|
| K417T | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 98% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| K417N | 45% | 96% | 98% | 100% | 0% | 0% | 93% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 8% | 0% | 0% | 0% | 0% | 0% |
| N439K | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 96% | 0% |
| N440K | 48% | 92% | 93% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| G446S | 48% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| L452R | 0% | 95% | 99% | 0% | 97% | 0% | 0% | 0% | 97% | 0% | 1% | 0% | 95% | 0% | 0% | 80% | 0% | 95% | 0% | 0% |
| L452Q | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 98% | 0% | 0% | 0% | 0% | 0% | 0% |
| S477N | 90% | 97% | 99% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 9% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 1% |
| T478K | 90% | 96% | 99% | 100% | 96% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 1% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| E484K | 0% | 0% | 0% | 0% | 0% | 0% | 91% | 95% | 0% | 97% | 72% | 100% | 0% | 0% | 97% | 0% | 14% | 0% | 0% | 13% |
| E484Q | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 93% | 0% | 0% | 80% | 0% | 0% | 0% | 0% |
| E484A | 90% | 97% | 99% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| F486V | 0% | 97% | 99% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| F490S | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 98% | 0% | 0% | 0% | 0% | 0% | 0% |
| Q493R | 91% | 0% | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| S494P | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| G496S | 89% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| Q498R | 90% | 95% | 99% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| N501Y | 90% | 97% | 99% | 100% | 0% | 98% | 91% | 90% | 0% | 0% | 0% | 0% | 0% | 0% | 98% | 0% | 0% | 0% | 0% | 0% |
| Y505H | 90% | 96% | 99% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| T547K | 98% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| A570D | 0% | 0% | 0% | 0% | 0% | 99% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| Q613H | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 100% | 0% | 0% | 0% |
| D614G | 100% | 100% | 100% | 100% | 100% | 100% | 99% | 100% | 100% | 99% | 100% | 99% | 98% | 100% | 99% | 100% | 0% | 99% | 99% | 100% |
| H655Y | 100% | 100% | 100% | 100% | 0% | 0% | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| Q677H | 0% | 0% | 0% | 0% | 0% | 1% | 0% | 0% | 1% | 98% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 1% | 0% |
| N679K | 100% | 99% | 100% | 100% | 0% | 0% | 0% | 2% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | |
| P681H | 100% | 100% | 100% | 100% | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 1% | 0% | 99% | 2% | 0% | 5% | 1% | 38% |
| P681R | 0% | 0% | 0% | 0% | 99% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 97% | 0% | 0% | 85% | 100% | 0% | 0% | 0% |
| A701V | 14% | 1% | 0% | 0% | 0% | 0% | 99% | 0% | 0% | 0% | 88% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
Table A2.
The sequences included in the reference database.
Table A2.
The sequences included in the reference database.
| Reference | Variant | Lineage | Reference | Variant | Lineage |
|---|
| NC_045512.2 | Reference genome | EPI_ISL_9009322 | Delta | AY.4.2 |
| MT358641.1 | Wild Type | EPI_ISL_2921532 | Lambda | C.37 |
| MW772455.1 | Wild Type | EPI_ISL_2957834 | Lambda | C.37 |
| MT470142.1 | Wild Type | EPI_ISL_2923197 | Lambda | C.37 |
| MT192772.1 | Wild Type | EPI_ISL_2930541 | Lambda | C.37 |
| EPI_ISL_833226 | Alpha | B.1.1.7 | EPI_ISL_2932985 | Lambda | C.37.1 |
| EPI_ISL_832204 | Alpha | B.1.1.7 | EPI_ISL_6640917 | Omicron | BA.1.17 |
| EPI_ISL_833038 | Alpha | B.1.1.7 | EPI_ISL_6670244 | Omicron | BA.1.17 |
| EPI_ISL_833147 | Alpha | B.1.1.7 | EPI_ISL_6699736 | Omicron | BA.1 |
| EPI_ISL_832169 | Alpha | B.1.1.7 | EPI_ISL_6699770 | Omicron | BA.1 |
| EPI_ISL_833043 | Alpha | B.1.1.7 | EPI_ISL_6704875 | Omicron | BA.1 |
| EPI_ISL_660606 | Beta | B.1.351 | EPI_ISL_11209238 | Omicron | XE |
| EPI_ISL_745123 | Beta | B.1.351 | EPI_ISL_11401887 | Omicron | XE |
| EPI_ISL_660608 | Beta | B.1.351 | EPI_ISL_11490660 | Omicron | XE |
| EPI_ISL_700587 | Beta | B.1.351 | EPI_ISL_11607013 | Omicron | XE |
| EPI_ISL_712081 | Beta | B.1.351 | EPI_ISL_11878447 | Omicron | XE |
| EPI_ISL_792680 | Gamma | P.1 | EPI_ISL_6795834 | Omicron | BA.2 |
| EPI_ISL_833170 | Gamma | P.1 | EPI_ISL_8053581 | Omicron | BA.2 |
| EPI_ISL_833169 | Gamma | P.1 | EPI_ISL_8166509 | Omicron | BA.2.10 |
| EPI_ISL_833167 | Gamma | P.1 | EPI_ISL_8253256 | Omicron | BA.2 |
| EPI_ISL_792681 | Gamma | P.1 | EPI_ISL_13399356 | Omicron | BA.5.1 |
| EPI_ISL_1004177 | Eta | B.1.525 | EPI_ISL_13399365 | Omicron | BA.5.1 |
| EPI_ISL_1018082 | Eta | B.1.525 | EPI_ISL_13399411 | Omicron | BA.5.1 |
| EPI_ISL_1020303 | Eta | B.1.525 | EPI_ISL_13399413 | Omicron | BA.5.1 |
| EPI_ISL_1024908 | Eta | B.1.525 | EPI_ISL_13399833 | Omicron | BA.5.1 |
| EPI_ISL_1035823 | Eta | B.1.525 | EPI_ISL_13402144 | Omicron | BA.4 |
| EPI_ISL_2726706 | Delta | AY.4 | EPI_ISL_13399316 | Omicron | BA.4 |
| EPI_ISL_2726793 | Delta | AY.75 | EPI_ISL_13399339 | Omicron | BA.4 |
| EPI_ISL_2727568 | Delta | AY.44 | EPI_ISL_13402674 | Omicron | BA.4.1.1 |
| EPI_ISL_2727864 | Delta | AY.46 | EPI_ISL_13405368 | Omicron | BA.4.1 |
| EPI_ISL_2727605 | Delta | AY.75 | | | |
References
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Kustin, T.; Stern, A. Biased Mutation and Selection in RNA Viruses. Mol. Biol. Evol. 2020, 38, 575–588. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of Spontaneous Mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Manzanares-Meza, L.D.; Medina-Contreras, O. SARS-CoV-2 and influenza: A comparative overview and treatment implications. Bol. Med. Hosp. Infant Mex. 2020, 77, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- ECDC-European Center for Disease Control. SARS-CoV-2 Variants of Concern as of 6 May 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 3 November 2022).
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Heal. Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 Variants, Vaccines, and Host Immunity. Front Immunol. 2022, 12, 809244. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med Virol. 2021, 94, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; Veinotte, K.; et al. SARS-CoV-2 Spike Protein Variant D614G Increases Infectivity and Retains Sensitivity to Antibodies That Target the Receptor Binding Domain. BioRxiv 2020, 2020, 187757. [Google Scholar] [CrossRef]
- Jang, M.; Park, Y.I.; Cha, Y.E.; Park, R.; Namkoong, S.; Lee, J.I.; Park, J. Tea Polyphenols EGCG and Theaflavin Inhibit the Activity of SARS-CoV-2 3CL-Protease in Vitro. Evid.-Based Complement. Altern. Med. 2020, 2020, 5630838. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, L. Human SARS-CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinform. 2021, 89, 569–576. [Google Scholar] [CrossRef]
- Xia, X. Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021, 21, 340–341. [Google Scholar] [CrossRef]
- Wang, H.; Jean, S.; Eltringham, R.; Madison, J.; Snyder, P.; Tu, H.; Jones, D.M.; Leber, A.L. Mutation-Specific SARS-CoV-2 PCR Screen: Rapid and Accurate Detection of Variants of Concern and the Identification of a Newly Emerging Variant with Spike L452R Mutation. J. Clin. Microbiol. 2021, 59, e0092621. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Ishige, T.; Fujikawa, T.; Miyabe, A.; Murata, S.; Kawasaki, K.; Nishimura, M.; Taniguchi, T.; Igari, H.; Matsushita, K. Development of multiplex S-gene-targeted RT-PCR for rapid identification of SARS-CoV-2 variants by extended S-gene target failure. Clin. Chim. Acta 2022, 536, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Murillo, E.; Palacio-Rua, K.; Afanador-Ayala, C.; García-Correa, J.F.; Zuluaga, A.F. Validación de una nueva estrategia para la identificación de variantes de SARS-CoV-2 mediante secuenciación del gen espiga por Sanger. Enferm. Infecc. Microbiol. Clin. 2022. [Google Scholar] [CrossRef]
- Matsubara, M.; Imaizumi, Y.; Fujikawa, T.; Ishige, T.; Nishimura, M.; Miyabe, A.; Murata, S.; Kawasaki, K.; Taniguchi, T.; Igari, H.; et al. Tracking SARS-CoV-2 variants by entire S-gene analysis using long-range RT-PCR and Sanger sequencing. Clin. Chim. Acta 2022, 530, 94–98. [Google Scholar] [CrossRef]
- Wagner, G.E.; Totaro, M.G.; Volland, A.; Lipp, M.; Saiger, S.; Lichtenegger, S.; Forstner, P.; von Laer, D.; Oberdorfer, G.; Steinmetz, I. A Novel High-Throughput Nanopore-Sequencing-Based Strategy for Rapid and Automated S-Protein Typing of SARS-CoV-2 Variants. Viruses 2021, 13, 2548. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martínez-Murcia, A.; Bru, G.; Navarro, A.; Ros-Tárraga, P.; García-Sirera, A.; Pérez, L. Comparative in silico design and validation of GPS™ COVID-19 dtec-RT-qPCR test. J. Appl. Microbiol. 2020, 130, 2–13. [Google Scholar] [CrossRef]
- Martínez-Murcia, A.; García-Sirera, A.; Navarro, A.; Pérez, L. Current RT-qPCR to detect SARS-CoV-2 may give positive results for related coronaviruses. Arch. Microbiol. 2022, 204, 1–6. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




{kind=link}
{kind=link}

