

The Impact of Deleting Stem-Loop 1 of Epstein–Barr Virus-Encoded RNA 1 on Cell Proliferation


Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of EBER-1 Stem-Loop Mutants, Cloning and Transfection
2.2. Cell Culture
2.3. Monitoring Cell Growth and Viability
2.4. Cell Cycle Analysis
2.5. Subcellular Fractionation, Nucleic Acid Isolation, qRT-PCR, and Microarray
2.6. Isolation of Total and Subcellular Proteins and Western Blotting
2.7. Statistical Analysis
3. Results
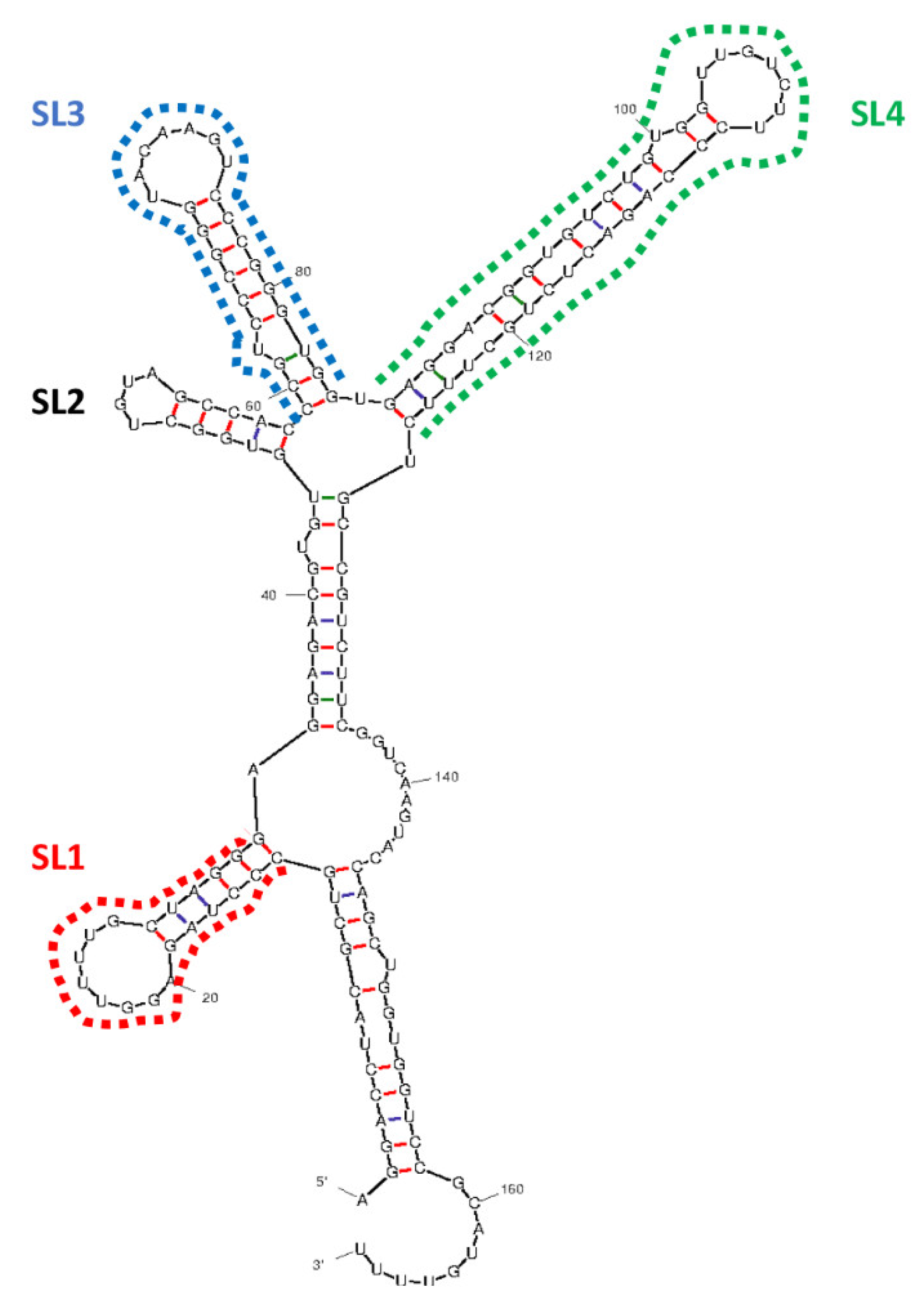
3.1. Prediction of RNA Structure, Confirmation of Deletion and Transfection, and Determination of Plasmid Copy Number
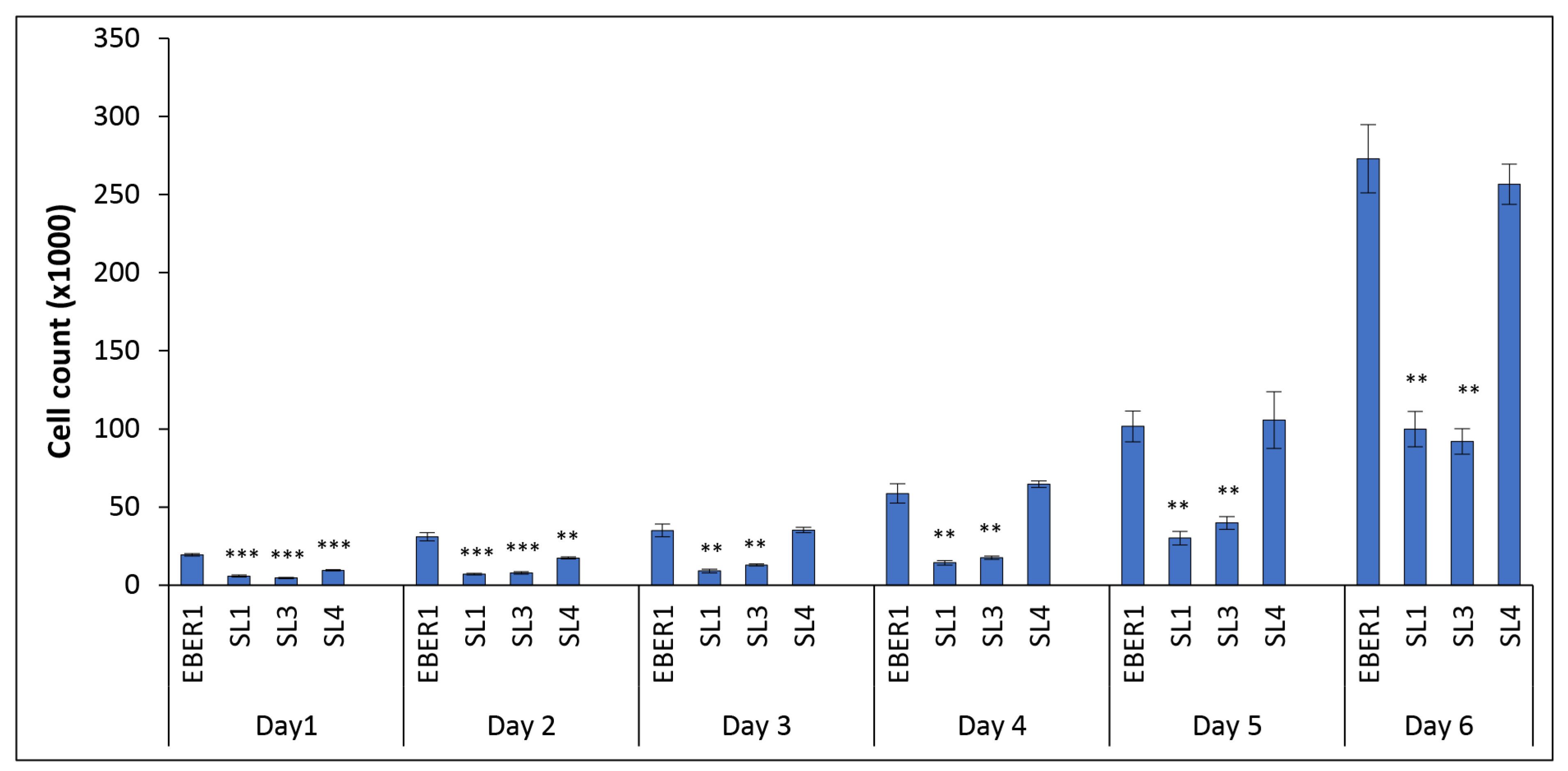
3.2. The Structure of EBER1 Is Important in Inducing Proliferation
3.3. EBER1-Induced Cell Proliferation Is Not Affected by Cell Death or DNA Repair
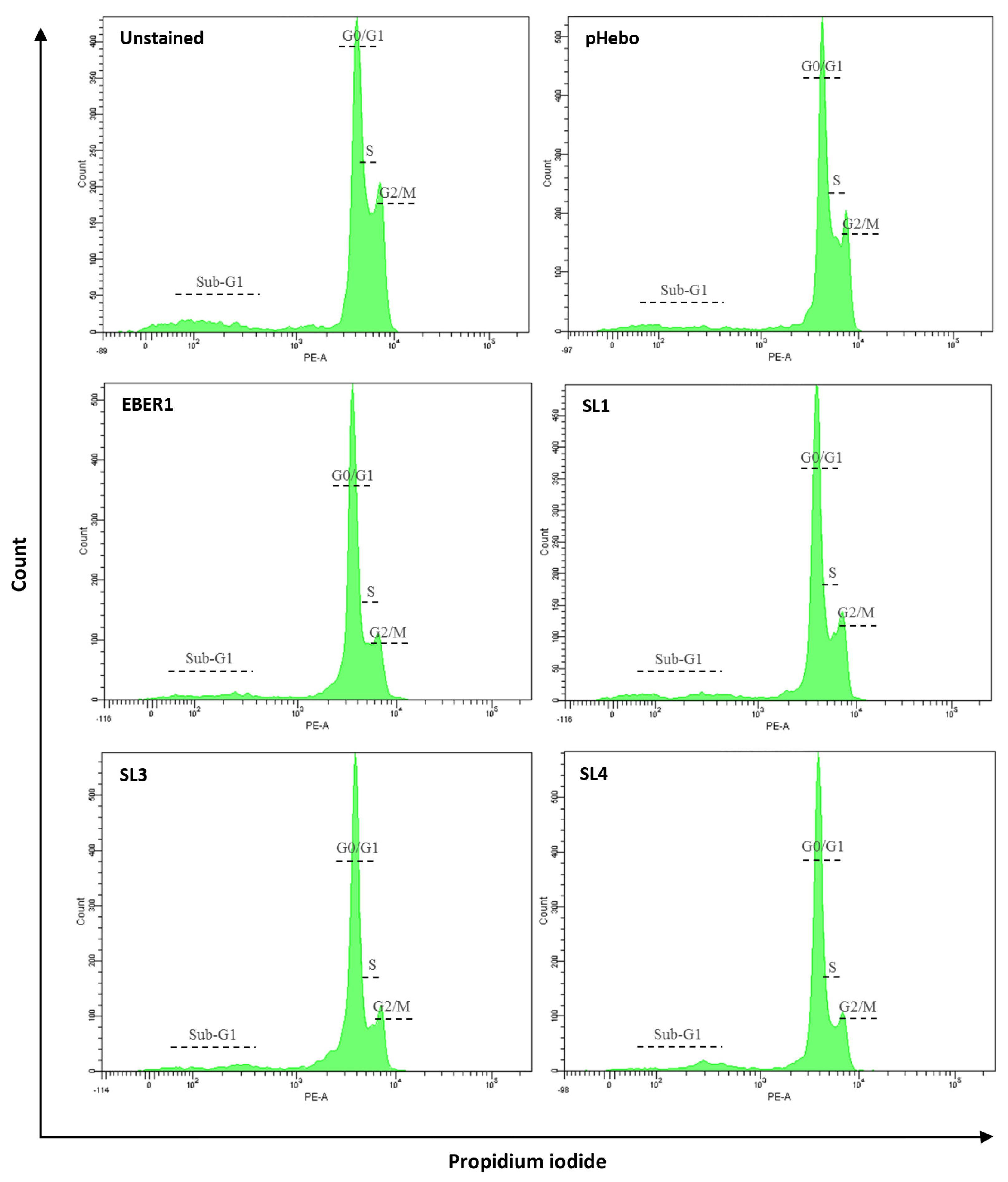
3.4. EBER1 Transfected Cells Are More in G2/M Phase
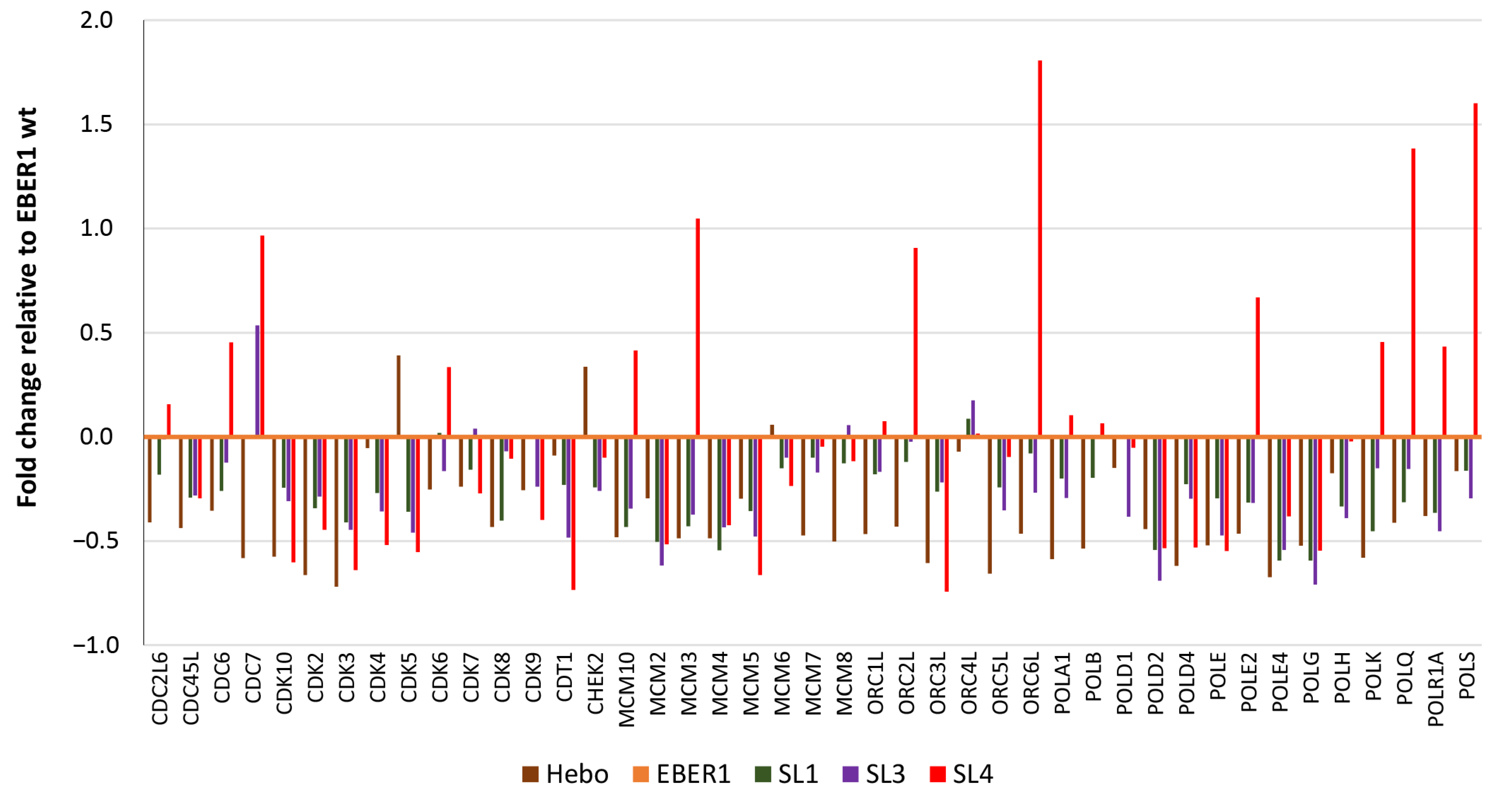
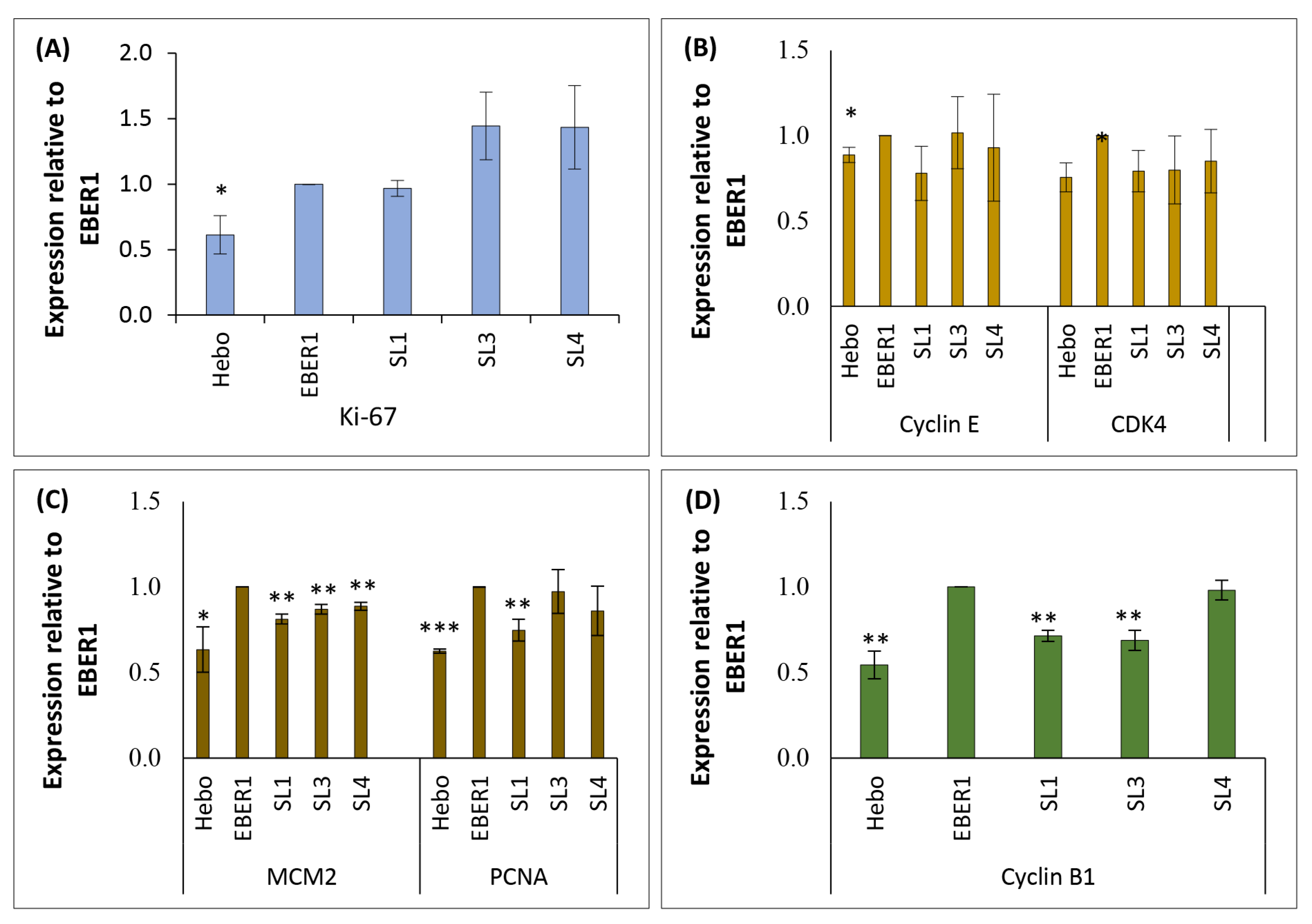
3.5. Genes Involved in Cell Cycle Progression Are Upregulated in Wildtype EBER1
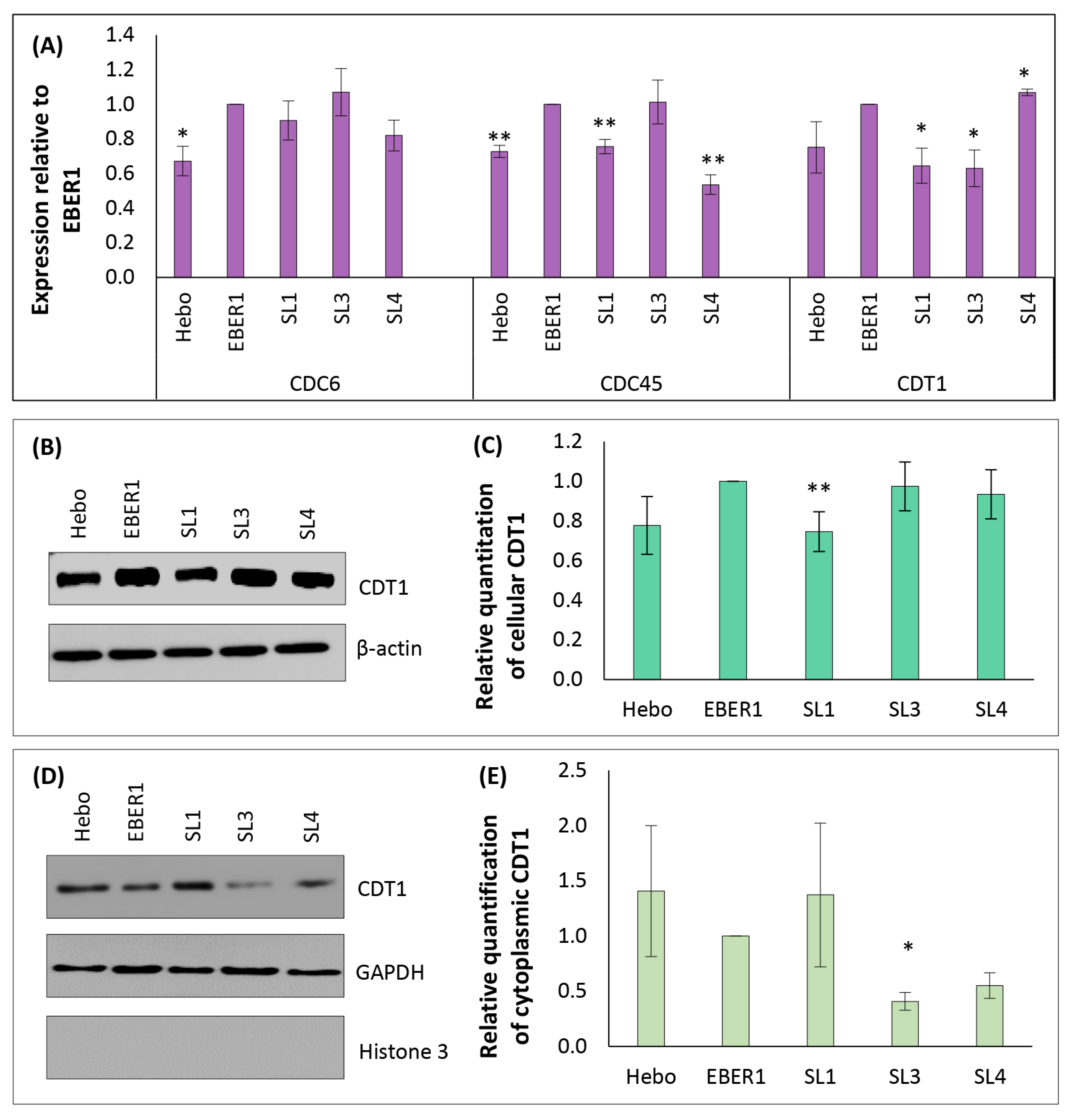
3.6. EBER1 Upregulates Genes Involved in Cell Cycle Progression and Proliferation
3.7. Mechanism of EBER1-Induced Cell Proliferation Appears to Involve CDT1 Upregulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, M.A.; De Matteo, E.; Gass, H.; Vazquez, P.M.; Lara, J.; Gonzalez, P.; Preciado, M.V.; Chabay, P.A. Characterization of Epstein Barr virus latency pattern in Argentine breast carcinoma. PLoS ONE 2010, 5, e13603. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Philip, P.S.; Tariq, S.; Khan, G. Epstein-Barr Virus-Encoded Small RNAs (EBERs) Are Present in Fractions Related to Exosomes Released by EBV-Transformed Cells. PLoS ONE 2014, 9, e99163. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Role of EBERs in the Pathogenesis of EBV Infection. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2010; Volume 107, pp. 119–136. [Google Scholar]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Katiyar-Agarwal, S.; Jin, H. Role of Small RNAs in Host-Microbe Interactions. Annu. Rev. Phytopathol. 2010, 48, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Eilebrecht, S.; Pellay, F.-X.; Odenwälder, P.; Brysbaert, G.; Benecke, B.-J.; Benecke, A. EBER2 RNA-induced transcriptome changes identify cellular processes likely targeted during Epstein Barr virus infection. BMC Res. Notes 2008, 1, 11–14. [Google Scholar] [CrossRef]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef]
- Herbert, K.M.; Pimienta, G. Consideration of Epstein-Barr Virus-Encoded Noncoding RNAs EBER1 and EBER2 as a Functional Backup of Viral Oncoprotein Latent Membrane Protein 1. MBio 2016, 7, e01926-15. [Google Scholar] [CrossRef]
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615–1630. [Google Scholar] [CrossRef]
- Yajima, M.; Kanda, T.; Takada, K. Critical Role of Epstein-Barr Virus (EBV)-Encoded RNA in Efficient EBV-Induced B-Lymphocyte Growth Transformation. J. Virol. 2005, 79, 4298–4307. [Google Scholar] [CrossRef]
- Yamamoto, N.; Takizawa, T.; Iwanaga, Y.; Shimizu, N.; Yamamoto, N. Malignant transformation of B lymphoma cell line BJAB by Epstein-Barr virus-encoded small RNAs. FEBS Lett. 2000, 484, 153–158. [Google Scholar] [CrossRef]
- Ahmed, W.; Hassan, Z.; Abdelmowla, Y.A.; Philip, P.S.; Shmygol, A.; Khan, G. Epstein-Barr virus noncoding small RNA (EBER1) induces cell proliferation by up-regulating cellular mitochondrial activity and calcium influx. Virus Res. 2021, 305, 198550. [Google Scholar] [CrossRef] [PubMed]
- Houmani, J.L.; Davis, C.I.; Ruf, I.K. Growth-promoting properties of Epstein-Barr virus EBER-1 RNA correlate with ribosomal protein L22 binding. J. Virol. 2009, 83, 9844–9853. [Google Scholar] [CrossRef] [PubMed]
- Samanta, M.; Takada, K. Modulation of innate immunity system by Epstein–Barr virus-encoded non-coding RNA and oncogenesis. Cancer Sci. 2010, 101, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.M. Advances in RNA secondary and tertiary structure analysis by chemical probing. Curr. Opin. Struct. Biol. 2010, 20, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Ishii, K.; Tahara, T.; Kitao, A. Origins of biological function in DNA and RNA hairpin loop motifs from replica exchange molecular dynamics simulation. Phys. Chem. Chem. Phys. 2018, 20, 2990–3001. [Google Scholar] [CrossRef]
- Heckman, K.L.; Pease, L.R. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2007, 2, 924–932. [Google Scholar] [CrossRef]
- Akhlaq, S.; Panicker, N.G.; Philip, P.S.; Ali, L.M.; Dudley, J.P.; Rizvi, T.A.; Mustafa, F. A cis-Acting Element Downstream of the Mouse Mammary Tumor Virus Major Splice Donor Critical for RNA Elongation and Stability. J. Mol. Biol. 2018, 430, 4307–4324. [Google Scholar] [CrossRef]
- Khan, G.; Coates, P.J.; Kangro, H.O.; Slavin, G. Epstein Barr virus (EBV) encoded small RNAs: Targets for detection by in situ hybridisation with oligonucleotide probes. J. Clin. Pathol. 1992, 45, 616–620. [Google Scholar] [CrossRef]
- Ahmed, W.; Tariq, S.; Khan, G. Tracking EBV-encoded RNAs (EBERs) from the nucleus to the excreted exosomes of B-lymphocytes. Sci. Rep. 2018, 8, 15438. [Google Scholar] [CrossRef]
- Ryan, J.L.; Fan, H.; Glaser, S.L.; Schichman, S.A.; Raab-Traub, N.; Gulley, M.L. Epstein-Barr Virus Quantitation by Real-Time PCR Targeting Multiple Gene Segments: A Novel Approach to Screen for the Virus in Paraffin-Embedded Tissue and Plasma. J. Mol. Diagn. 2004, 6, 378–385. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Hassan, Z.; Mustafa, S.; Rahim, R.A.; Isa, N.M. Anti-breast cancer effects of live, heat-killed and cytoplasmic fractions of Enterococcus faecalis and Staphylococcus hominis isolated from human breast milk. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Bender, T.P. Identification of newly transcribed RNA. Curr. Protoc. Mol. Biol. 2007, 4, 4.10. [Google Scholar] [CrossRef]
- Shimizu, A.; Nakatani, Y.; Nakamura, T.; Jinno-Oue, A.; Ishikawa, O.; Boeke, J.D.; Takeuchi, Y.; Hoshino, H. Characterisation of cytoplasmic DNA complementary to non-retroviral RNA viruses in human cells. Sci. Rep. 2014, 4, 5074. [Google Scholar] [CrossRef] [PubMed]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]
- Glickman, J.N.; Howe, J.G.; Steitz, J.A. Structural Analyses of EBERI and EBER2 Ribonucleoprotein Particles Present in Epstein-Barr Virus-Infected Cells. J. Virol. 1988, 62, 902–911. [Google Scholar] [CrossRef]
- Zembruski, N.C.; Stache, V.; Haefeli, W.; Weiss, J. 7-Aminoactinomycin D for apoptosis staining in flow cytometry. Anal. Biochem. 2012, 429, 79–81. [Google Scholar] [CrossRef]
- DaSilva, L. Functional Characterization of the Origin Recognition Complex (ORC) in S. cerevisiae. Master’s Thesis, University of Waterloo, Waterloo, ON, Canada, 2008. Available online: http://hdl.handle.net/10012/3518 (accessed on 9 October 2022).
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The Interplay Between Epstein Barr Virus and B Lymphocytes: Implications for Infection, Immunity, and Disease. Immunol. Res. 2014, 58, 268. [Google Scholar] [CrossRef]
- Lin, Z.; Swan, K.; Zhang, X.; Cao, S.; Brett, Z.; Drury, S.; Strong, M.J.; Fewell, C.; Puetter, A.; Wang, X.; et al. Secreted Oral Epithelial Cell Membrane Vesicles Induce Epstein-Barr Virus Reactivation in Latently Infected B Cells. J. Virol. 2016, 90, 3469–3479. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. Epstein–Barr virus replicating in epithelial cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16242–16243. [Google Scholar] [CrossRef] [PubMed]
- Dugan, J.P.; Coleman, C.B.; Haverkos, B. Opportunities to target the life cycle of Epstein-Barr Virus (EBV) in EBV-associated lymphoproliferative disorders. Front. Oncol. 2019, 9, 127. [Google Scholar] [CrossRef]
- Lee, N.; Steitz, J.A. Noncoding RNA-guided recruitment of transcription factors: A prevalent but undocumented mechanism? BioEssays 2015, 37, 936–941. [Google Scholar] [CrossRef]
- Lee, N.; Moss, W.N.; Yario, T.A.; Steitz, J.A. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 2015, 160, 607–618. [Google Scholar] [CrossRef]
- Gregorovic, G.; Bosshard, R.; Karstegl, C.E.; White, R.E.; Pattle, S.; Chiang, A.K.S.; Dittrich-Breiholz, O.; Kracht, M.; Russ, R.; Farrell, P.J. Cellular gene expression that correlates with EBER expression in Epstein-Barr virus-infected lymphoblastoid cell lines. J. Virol. 2011, 85, 3535–3545. [Google Scholar] [CrossRef]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA Viruses and Forty-One Hosts: Viral Small RNAs and Modulation of Small RNA Repertoires in Vertebrate and Invertebrate Systems. PLoS Pathog. 2010, 6, e1000764. [Google Scholar] [CrossRef] [PubMed]
- Repellin, C.E.; Tsimbouri, P.M.; Philbey, A.W.; Wilson, J.B. Lymphoid hyperplasia and lymphoma in transgenic mice expressing the small non-coding RNA, EBER1 of Epstein-Barr virus. PLoS ONE 2010, 5, e9092. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.; Vyas, J.; Laing, K.G.; Clemens, M.J. Ribosomal protein L22 inhibits regulation of cellular activities by the Epstein-Barr virus small RNA EBER-1. Eur. J. Biochem. 2004, 271, 1895–1905. [Google Scholar] [CrossRef]
- Toczyski, D.P.; Matera, A.G.; Ward, D.C.; Steitz, J.A. The Epstein–Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 3463–3467. [Google Scholar] [CrossRef]
- Fok, V.; Mitton-Fry, R.M.; Grech, A.; Steitz, J.A. Multiple domains of EBER 1, an Epstein-Barr virus noncoding RNA, recruit human ribosomal protein L22. RNA 2006, 12, 872–882. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.S.; Pal, A.D.; Banerjee, S. Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology 2013, 443, 294–305. [Google Scholar] [CrossRef]
- Kipreos, E.T.; Heuvel, S.V.D. Developmental Control of the Cell Cycle: Insights from Caenorhabditis elegans. Genetics 2019, 211, 797–829. [Google Scholar] [CrossRef]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % G1 | % S | % G2/M | |
|---|---|---|---|
| Hebo | 43.4 | 26.4 | 34.0 |
| EBER1 | 30.9 | 27.8 | 43.0 |
| SL1 | 36.5 | 25.3 | 41.5 |
| SL3 | 45.8 | 26.5 | 35.9 |
| SL4 | 45.6 | 30.9 | 30.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, Z.; Philip, P.S.; Khan, G. The Impact of Deleting Stem-Loop 1 of Epstein–Barr Virus-Encoded RNA 1 on Cell Proliferation. Viruses 2022, 14, 2538. https://doi.org/10.3390/v14112538
Hassan Z, Philip PS, Khan G. The Impact of Deleting Stem-Loop 1 of Epstein–Barr Virus-Encoded RNA 1 on Cell Proliferation. Viruses. 2022; 14(11):2538. https://doi.org/10.3390/v14112538
Chicago/Turabian StyleHassan, Zubaida, Pretty S. Philip, and Gulfaraz Khan. 2022. "The Impact of Deleting Stem-Loop 1 of Epstein–Barr Virus-Encoded RNA 1 on Cell Proliferation" Viruses 14, no. 11: 2538. https://doi.org/10.3390/v14112538
APA StyleHassan, Z., Philip, P. S., & Khan, G. (2022). The Impact of Deleting Stem-Loop 1 of Epstein–Barr Virus-Encoded RNA 1 on Cell Proliferation. Viruses, 14(11), 2538. https://doi.org/10.3390/v14112538