
Occurrence and Molecular Variability of the Main Kiwifruit Viruses in the Sichuan Province of China
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Detection of Viruses by Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
2.3. Cloning and Sequencing
2.4. Calculation of the Mosaic Area and Disease Severity Classification
2.5. Recombination and Phylogenetic Analysis
2.6. Genetic Diversity, Genetic Differentiation of the Populations, and Gene Flow
2.7. Analysis of the Selection Pressure and Population Demographics
2.8. Comparison of the Whole AcCRaV Genome
2.9. Statistical Analysis
3. Results
3.1. Virus Detection and Disease Index
3.2. Symptoms of Kiwifruit Viruses
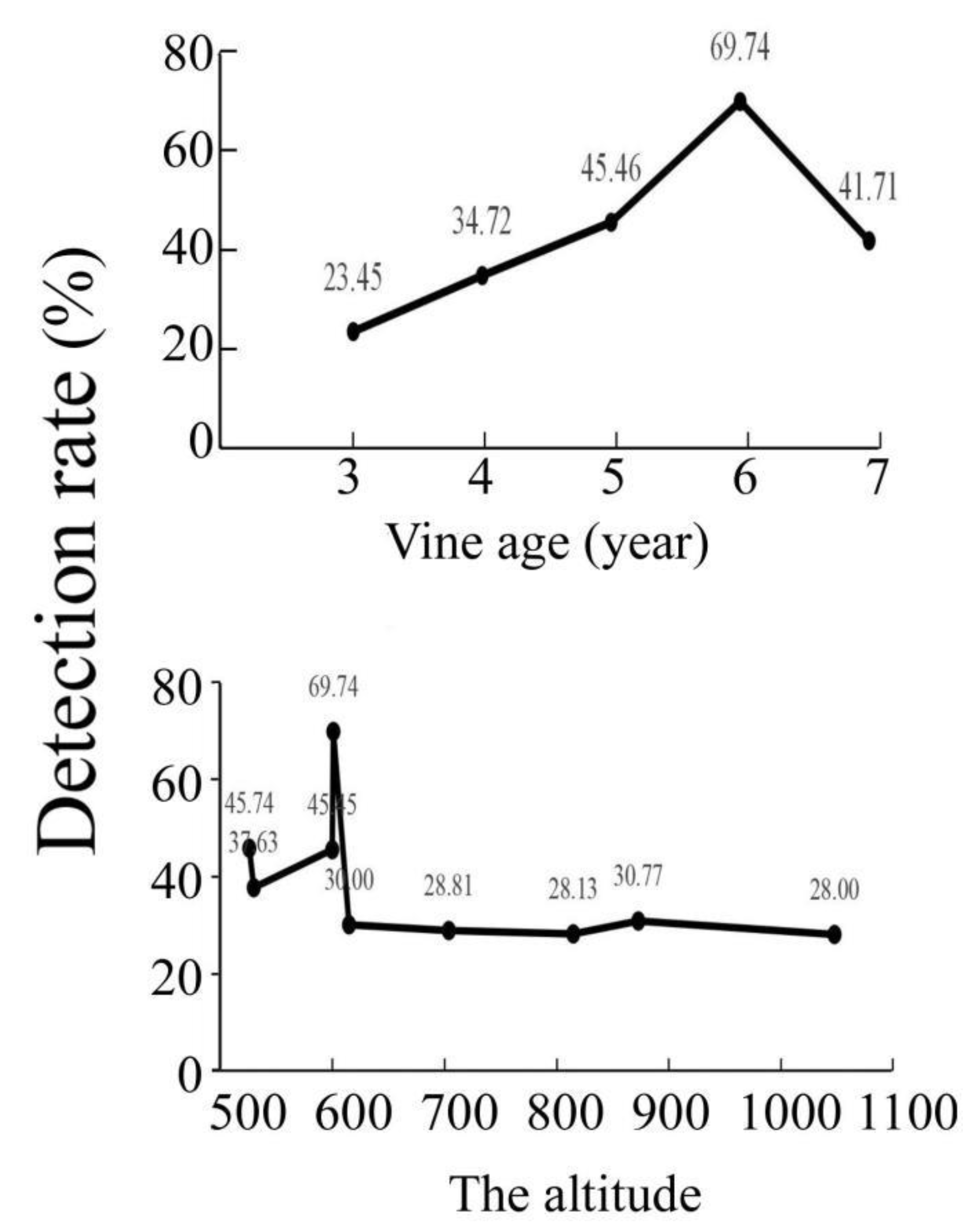
3.3. The Detection Rate Was Affected by Vine Age and Altitude
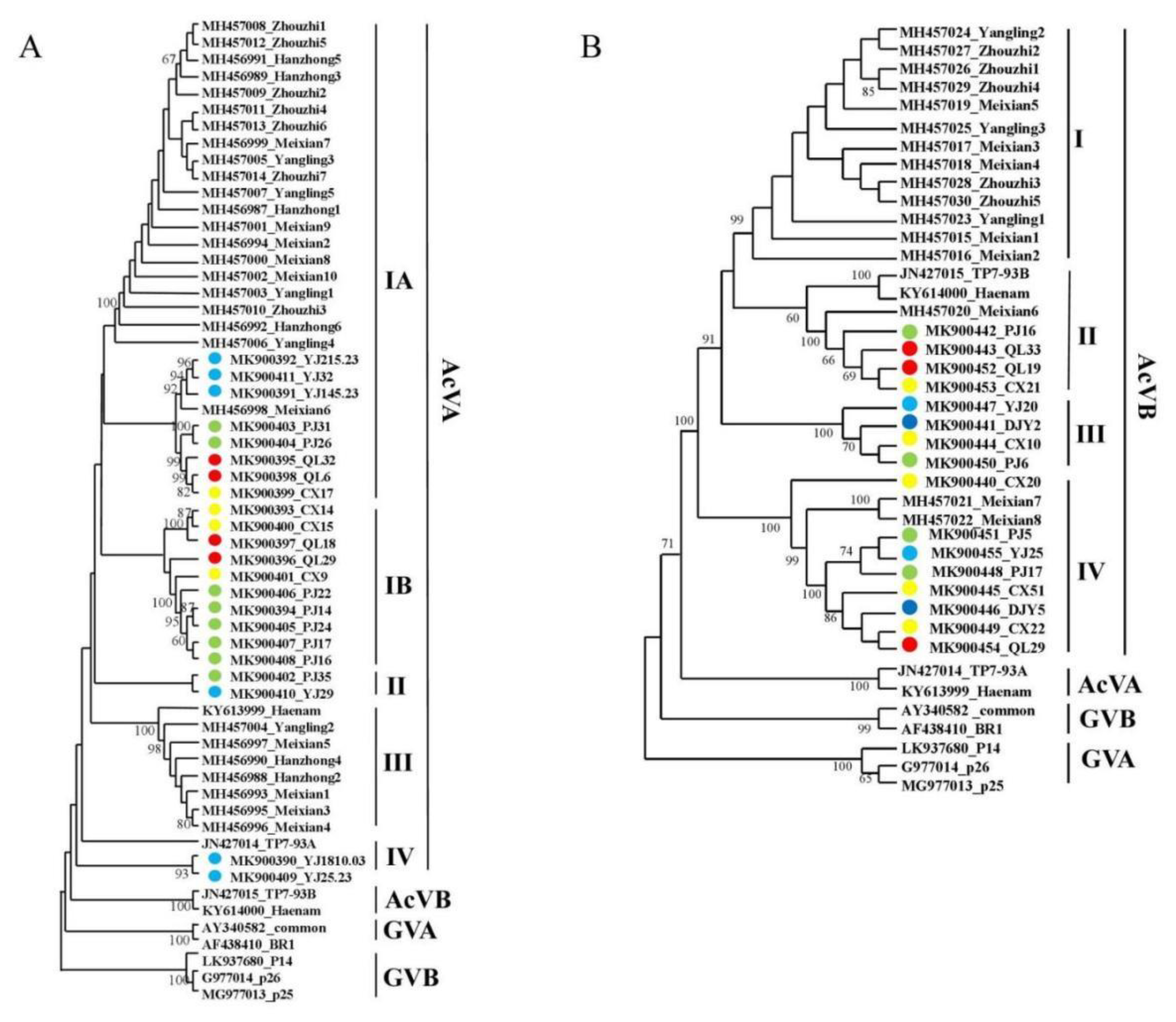
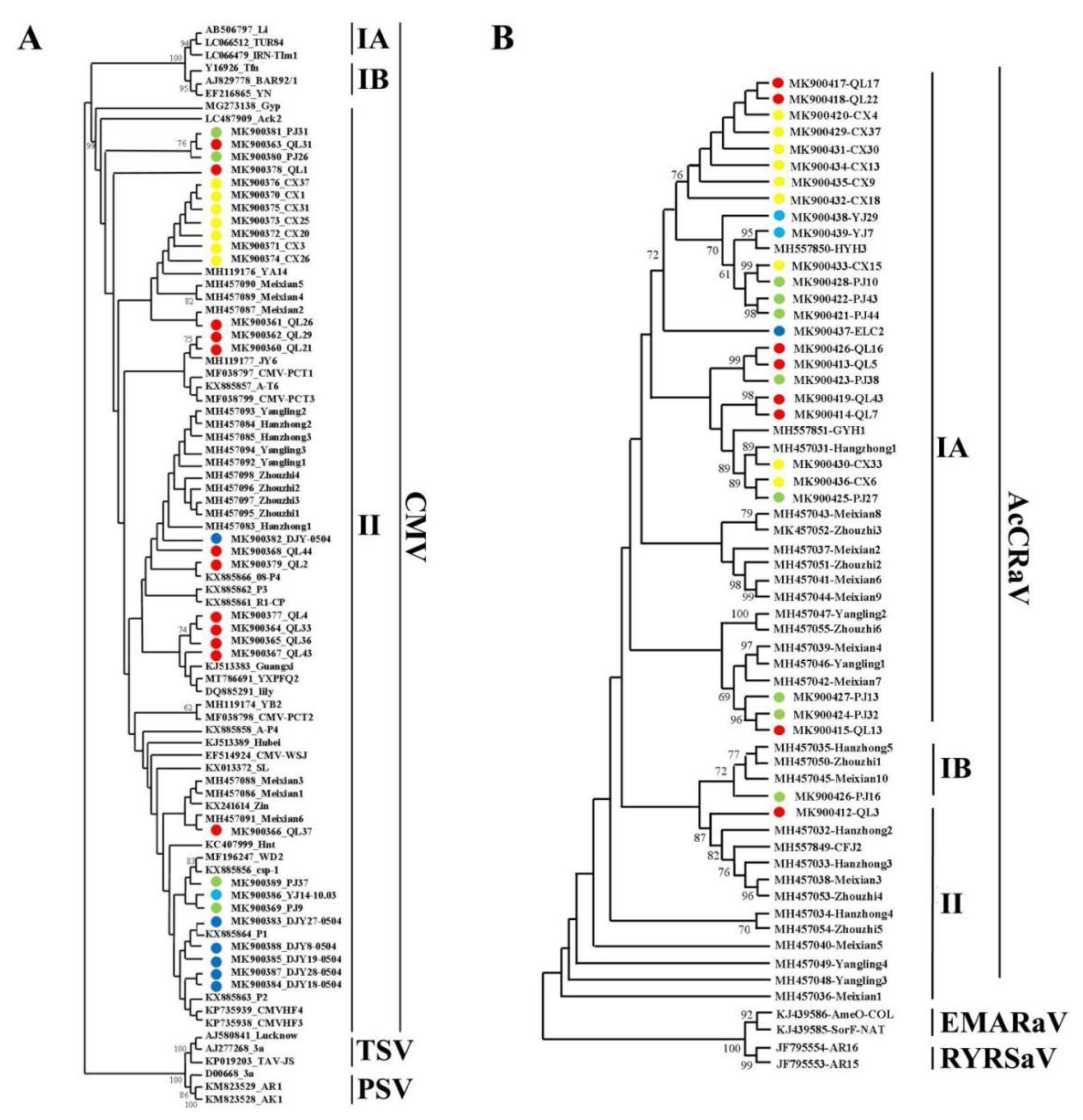
3.4. Population Genetic Analysis of Viral Coat Protein Sequences
3.5. Genetic Diversity Analysis of Kiwifruit Virus CP Genes
3.6. Population Differentiation and Gene Flow
3.7. Selection Pressure
3.8. Analysis of Population Dynamics
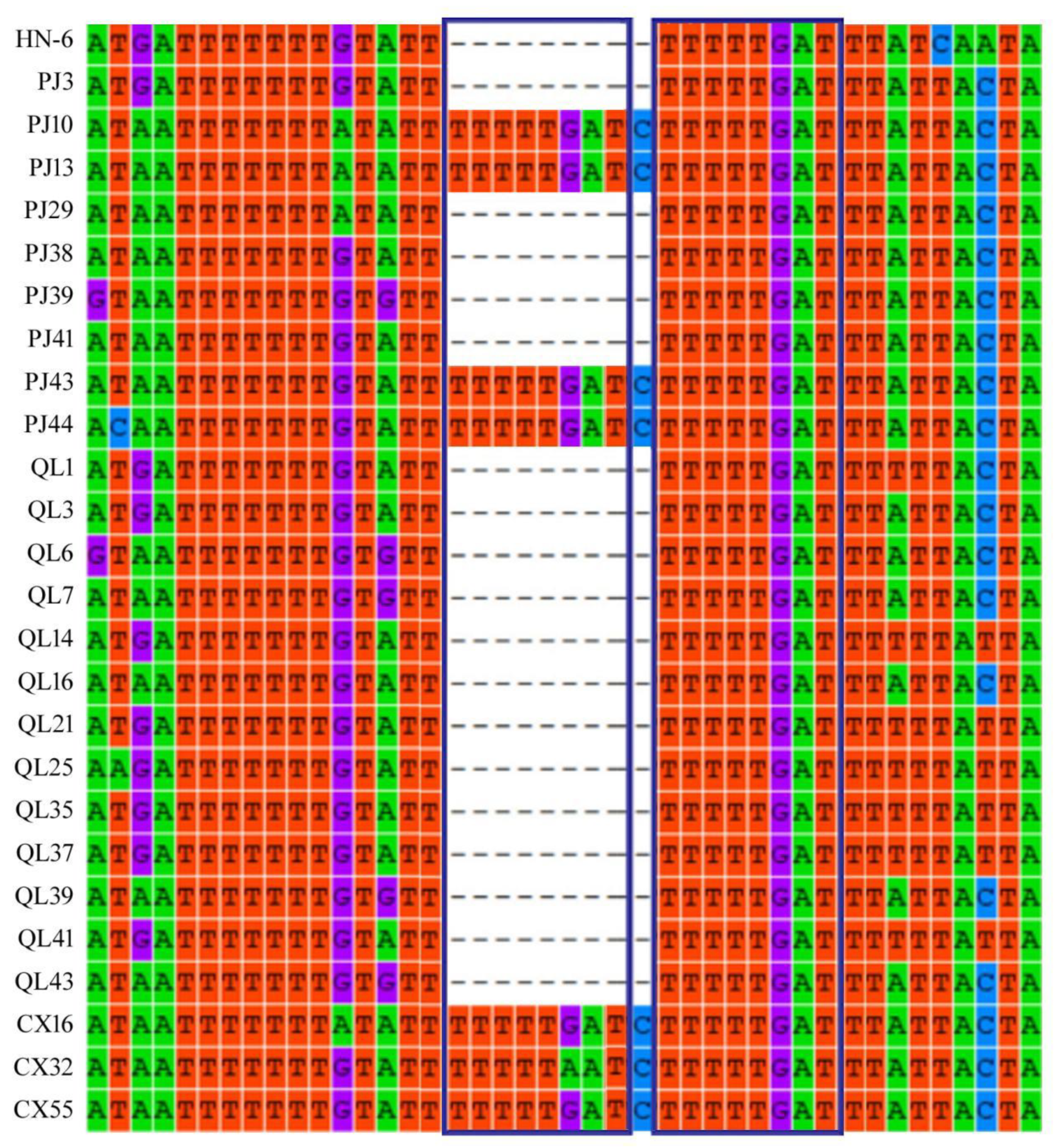
3.9. Repeated Sequence Analysis of the 5′-Terminal Non-Coding Region of RNA3 in AcCRaV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Wang, G.; Bai, J.; Zhang, Y.; Wang, Y.; Wen, S.; Li, L.; Yang, Z.; Hong, N. A novel Actinidia cytorhabdovirus characterized using genomic and viral protein interaction features. Mol. Plant Pathol. 2021, 22, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.G.; Pearson, M.N.; Chavan, R.R.; Woo, E.N.Y.; Lebas, B.S.M.; Veerakone, S.; Ratti, C.; Biccheri, R.; MacDiarmid, R.M.; Cohen, D. Viruses of Kiwifruit (Actinidia species). J. Plant Pathol. 2013, 99, 221–235. [Google Scholar]
- Blouin, A.G.; Chavan, R.R.; Pearson, M.N.; Macdiarmid, R.M.; Cohen, D. Detection and characterisation of two novel vitiviruses infecting Actinidia. Arch. Virol. 2012, 157, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Kim, H.; Yi, S.I. First Report of Actinidia virus A and B Infecting Actinidia chinensis in South Korea. Plant Dis. 2017, 101, 1560. [Google Scholar] [CrossRef]
- Stella, V.; Lia, W.; Liefting, J.T. The complete nucleotide sequence and genome organisation of a novel member of the family Betaflexiviridae from Actinidia chinensis. Arch. Virol. 2018, 163, 1367–1370. [Google Scholar]
- Zheng, Y.Z.; Wang, G.P.; Zhou, J.F.; Zhu, C.X.; Wang, L.P.; Xu, W.X.; Hong, N. The detection of Actinidia virus A and Actinidia virus B by RT–PCR and their molecular variation analysis. Acta. Hortic. Sin. 2015, 42, 665–671. [Google Scholar]
- Hernández-Rodríguez, L.; Pérez-Castro, J.M.; García-García, G.; Ramos-González, P.L.; Zamora-Rodríguez, V.; Ferriol-Marchena, X.; Peña-Bárzaga, I.; Batista-Le Riverend, L. Citrus leaf blotch virus in Cuba: First report and partial molecular characterization. Trop. Plant Pathol. 2016, 41, 147–154. [Google Scholar] [CrossRef]
- Zhu, C.X.; Wang, G.P.; Zheng, Y.Z.; Yang, Z.K.; Wang, L.P.; Xu, W.-X.; Hong, N. RT–PCR detection and sequence analysis of coat protein gene of Citrus leaf blotch virus infecting kiwifruit trees. Acta Phytopathol. Sin. 2016, 46, 11–16. [Google Scholar]
- Liu, H.; Song, S.; Wu, W.; Mi, W.L.; Shen, C.; Bai, B.X.; Wu, Y.F. Distribution and molecular characterization of Citrus leaf blotch virus from Actinidia in Shaanxi province, China. Eur. J. Plant Pathol. 2019, 154, 855–862. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Navarro, B.; Wang, G.P.; Wang, Y.X.; Yang, Z.K.; Xu, W.X.; Zhu, C.X.; Wang, L.P.; Di, F.; Hong, S.N. Actinidia chlorotic ringspot–associated virus: A novel emaravirus infecting kiwifruit plants. Mol. Plant Pathol. 2017, 18, 569–581. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zhai, L.F.; Wen, S.H.; Yang, Z.K.; Wang, G.P.; Hong, N. Molecular characterization of a novel Emaravrius infecting Actinidia spp. in China. Virus Res. 2020, 275, 197736. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kondo, H.; Andika, I.B.; Andika, I.B.; Liu, P.; Sun, L.; Wu, Y. Identification of genome recombination among Apple stem pitting isolates. J. Plant Pathol. 2016, 98, 595–601. [Google Scholar]
- Marais, A.; Faure, C.; Mustafayev, E.; Candresse, T. Characterization of new isolates of Apricot vein clearing–associated virus and of a new Prunus–infecting virus: Evidence for recombination as a driving force in Betaflexiviridae evolution. PLoS ONE 2015, 10, e0129469. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.G.; Biccheri, R.; Khalifa, M.E.; Pearson, M.N.; Poggi Pollini, C.; Hamiaux, C.; Cohen, D.; Ratti, C. Characterization of Actinidia virus 1, a new member of the family Closteroviridae encoding a thaumatin–like protein. Arch. Virol. 2017, 163, 229–234. [Google Scholar] [PubMed]
- Wang, D.; Liu, X.X.; Li, T.T.; Li, Q.; Liu, Y.; Gong, G.S.; Yang, H. First report of Cucumber mosaic virus infection in kiwifruit (Actinidia chinensis) in China. Plant Dis. 2018, 102, 1180. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, H.; Yang, Z.; Wen, L.; Wang, G.; Hong, N. Molecular characterization of an Apple stem grooving virus isolate from kiwifruit (Actinidia chinensis) in China. Can. J. Plant Pathol. 2018, 40, 76–83. [Google Scholar] [CrossRef]
- Pooja, B.; Vipin, H. Occurrence of Apple stem grooving virus on Rubus ellipticus, a perennial weed in India. Eur. J. Plant Pathol. 2018, 153, 311–319. [Google Scholar]
- Zhao, L.; Yang, W.; Zhang, Y.; Wu, Z.; Wang, Q.C.; Wu, Y. Occurrence and molecular variability of kiwifruit viruses in Actinidia deliciosa ‘Xuxiang’ in the Shaanxi province of China. Plant Dis. 2019, 103, 1309–1318. [Google Scholar]
- Pearson, M.N.; Cohen, D.; Chavan, R.; Blouin, A. Actinidia is a natural host to a wide range of plant viruses. Acta Hortic. 2010, 913, 467–471. [Google Scholar] [CrossRef]
- Bol, J.F. Alfalfa mosaic virus: Coat protein-dependent initiation of infection. Mol. Plant Pathol. 2003, 4, 1–8. [Google Scholar] [CrossRef]
- Hull, R. Alfalfa mosaic virus. Adv. Virus Res. 1969, 15, 365–433. [Google Scholar] [PubMed]
- Wang, Y.; Yang, Z.; Wang, G.; Yang, Z.K.; Wang, L.P.; Li, L. First report of the Tospovirus Tomato necrotic spot associated virus infecting kiwifruit (Actinidia sp.) in China. Plant Dis. 2016, 100, 2539–2540. [Google Scholar] [CrossRef]
- Von Bargen, S.; Langer, J.; Robel, J.; Rumbou, A.; Büttner, C. Complete nucleotide sequence of Cherry leaf roll virus (CLRV), a subgroup C nepovirus. Virus Res. 2012, 163, 678–683. [Google Scholar] [CrossRef]
- Rebenstorf, K.; Candresse, T.; Dulucq, M.J.; Büttner, C.; Obermeier, C. Host species–dependent population structure of a pollen–borne plant virus, Cherry leaf roll virus. J. Virol. 2006, 80, 2453–2462. [Google Scholar] [CrossRef]
- Woo, E.N.Y.; Clover, G.R.G.; Pearson, M.N. First report of Cherry leaf roll virus (CLRV) in Malus domestica. Australas. Plant Dis. Notes 2012, 7, 151–156. [Google Scholar] [CrossRef]
- Pasin, F.; Menzel, W.; Daròs, J.A. Harnessed viruses in the age of metagenomics and synthetic biology: An update on infectious clone assembly and biotechnologies of plant viruses. Plant Biotech. J. 2019, 17, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of plant viruses and disease management: Relevance of genetic diversity and evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef]
- Azzam, O.; Yambao, M.L.M.; Muhsin, M.; McNally, K.L.; Umadhay, K.M.L. Genetic diversity of rice tungro spherical virus in tungro–endemic provinces of the Philippines and Indonesia. Arch. Virol. 2000, 145, 1183–1197. [Google Scholar] [CrossRef]
- Garcíaarenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157. [Google Scholar] [CrossRef]
- Abdalla, O.A.; Ali, A. Genetic variability and evidence of a new subgroup in Watermelon mosaic virus Isolates. Pathogens 2021, 10, 1245. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo–Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Moradi, Z. Meta-transcriptomic analysis reveals an isolate of aphid lethal paralysis virus from Wisteria sinensis in Iran. Virus Res. 2022, 315, 198770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shang, J.; Jia, Q.; Li, K.; Yang, H.; Liu, H.H.; Tang, Z.Q.; Chang, X.L.; Zhang, M.; Wang, W.M.; et al. Genetic evolutionary analysis of soybean mosaic virus populations from three geographic locations in China based on the P1 and CP genes. Arch. Virol. 2019, 164, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Q.; Shang, J.; Zhang, L.; Du, D.B.; Yang, H.; Zeng, S.H.; Li, P.L.; Bawa, G.; Yu, L.; Hou, X.X.; et al. Characterization of synergy between Cucumber mosaic virus and Alternaria alternata in Nicotiana tabacum. Physiol. Mol. Plant Pathol. 2019, 108, 101404. [Google Scholar] [CrossRef]
- Waweru, B.W.; Miano, D.W.; Kilalo, D.C.; Rukundo, P.; Kimenju, J.W. Detection and distribution of viruses infecting hot pepper (Capsicum spp.) in Rwanda. J. Plant Pathol. 2021, 103, 573–585. [Google Scholar] [CrossRef]
- Mauck, K.E.; Kenney, J.; Chesnais, Q. Progress and challenges in identifying molecular mechanisms underlying host and vector manipulation by plant viruses. Curr. Opin. Insect. Sci. 2019, 33, 7–18. [Google Scholar] [CrossRef]
- Alomar, A.A.; Alto, B.W. Temperature-Mediated Effects on Mayaro Virus Vector Competency of Florida Aedes aegypti Mosquito Vectors. Viruses 2022, 14, 880. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, W.; Luo, C.; Li, Y.; Liu, T. Transmission comparisons of cucumber mosaic virus subgroup I and II isolates by different aphid species. J. Phytopathol. 2012, 160, 299–303. [Google Scholar] [CrossRef]
- Chavan, R.R.; Cohen, D.; Blouin, A.G.; Pearson, M.N. Characterization of the complete genome of ribgrass mosaic virus isolated from Plantago major L. from New Zealand and Actinidia spp. from China. Arch. Virol. 2012, 157, 1253–1260. [Google Scholar] [CrossRef]
- Dogimont, C.; Chovelon, V.; Pauquet, J.; Boualem, A.; Bendahmane, A. The Vat locus encodes for a CC-NBS-LRR protein that confers resistance to Aphis gossypii infestation and A. gossypii-mediated virus resistance. Plant J. 2014, 80, 993–1004. [Google Scholar] [CrossRef]
- Moradi, Z.; Mehrvar, M. Genetic variability and molecular evolution of Bean common mosaic virus populations in Iran: Comparison with the populations in the world. Eur. J. Plant Pathol. 2019, 154, 673–690. [Google Scholar] [CrossRef]
- Cunniffe, N.J.; Taylor, N.P.; Hamelin, F.M.; Jeger, M.J. Epidemiological and ecological consequences of virus manipulation of host and vector in plant virus transmission. PLoS Comput. Biol. 2021, 17, e1009759. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Mosaic Area ×106 (ppi) | Total Area ×106 (ppi) | Mosaic Rate | Disease Index (DAI) | Detection Rate |
|---|---|---|---|---|---|
| AcVA | 0.95 b | 5.22 a | 17.07% a | 5.57% | 9.03% |
| AcVB | 1.15 b | 5.49 a | 24.3% a | 3.94% | 7.10% |
| AcCRaV | 2.12 ab | 4.97 a | 42.4% a | 5.47% | 41.94% |
| CMV | 2.92 a | 6.20 a | 47.86% a | 7.92% | 9.68% |
| AcVA + AcVB | 0.25 a | 5.55 a | 4.868% a | 2.58% | 5.16% |
| AcVA + AcCRaV | 1.24 a | 5.78 a | 22.85% a | 2.79% | 7.10% |
| AcVB + AcCRaV | 0.05 a | 6.36 a | 0.75% b | 1.02% | 1.29% |
| AcVA + CMV | 1.59 a | 5.91 a | 25.63% a | 0.90% | 1.29% |
| AcVB + CMV | 1.51 a | 6.00 a | 24.41% a | 1.10% | 1.29% |
| AcCRaV + CMV | 1.96 a | 4.91 a | 40.63% a | 3.75% | 8.39% |
| AcVA + AcVB + AcCRaV | 2.04 b | 5.76 a | 34.08% b | 2.94% | 3.87% |
| AcVA + AcCRaV + CMV | 1.87 b | 5.58 a | 35.49% b | 0.90% | 1.29% |
| AcVB + AcCRaV + CMV | 2.56 ab | 5.05 a | 51.49% ab | 1.49% | 1.94% |
| AcVA + AcVB + AcCRaV + CMV | 4.70 a | 6.49 a | 72.42% a | 0.65% | 0.65% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, J.; Jia, Q.; Zhang, L.; Zhang, S.; Du, J.; Wang, W.; Shui, J. Occurrence and Molecular Variability of the Main Kiwifruit Viruses in the Sichuan Province of China. Viruses 2022, 14, 2460. https://doi.org/10.3390/v14112460
Shang J, Jia Q, Zhang L, Zhang S, Du J, Wang W, Shui J. Occurrence and Molecular Variability of the Main Kiwifruit Viruses in the Sichuan Province of China. Viruses. 2022; 14(11):2460. https://doi.org/10.3390/v14112460
Chicago/Turabian StyleShang, Jing, Qi Jia, Lei Zhang, Siqi Zhang, Junbo Du, Wenming Wang, and Jing Shui. 2022. "Occurrence and Molecular Variability of the Main Kiwifruit Viruses in the Sichuan Province of China" Viruses 14, no. 11: 2460. https://doi.org/10.3390/v14112460
APA StyleShang, J., Jia, Q., Zhang, L., Zhang, S., Du, J., Wang, W., & Shui, J. (2022). Occurrence and Molecular Variability of the Main Kiwifruit Viruses in the Sichuan Province of China. Viruses, 14(11), 2460. https://doi.org/10.3390/v14112460