
Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine
{kind=link}
Abstract
1. Introduction
2. Viral Metagenomics and Blood Transfusion
3. The Human Blood Virome
3.1. The Commensal Blood Donor Virome
3.2. In Search of Pathogenic Transfusion-Transmitted Viruses: Metagenomics Applied on Blood Donors Reporting Postdonation Illness
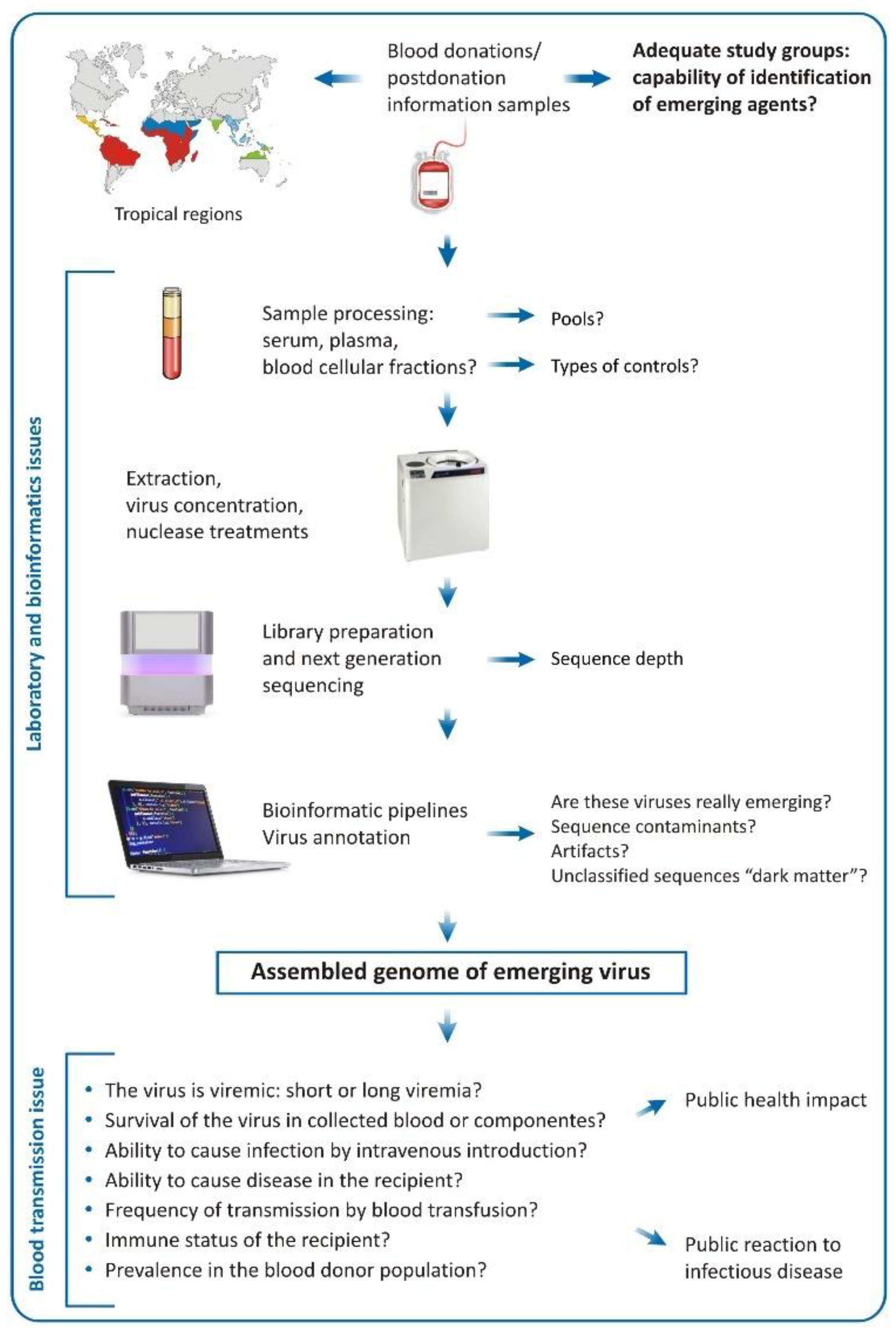
4. Challenges of the Metagenomic Analysis in Regards of Blood Donation
5. Final Remarks and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohsin, H.; Asif, A.; Fatima, M.; Rehman, Y. Potential role of viral metagenomics as a surveillance tool for the early detection of emerging novel pathogens. Arch. Microbiol. 2021, 203, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Chen, Y.M.; Wang, W.; Qin, X.C.; Holmes, E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Liu, L.; Huang, X.; Ma, H.; Zhang, Y.; Du, Y.; Wang, P.; Tang, X.; Wang, H.; Kang, K.; et al. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: Discovery of a new bunyavirus. PLoS Pathog. 2011, 7, e1002369. [Google Scholar] [CrossRef]
- de Mello Malta, F.; Amgarten, D.; Nastri, A.C.S.S.; Ho, Y.L.; Boas Casadio, L.V.; Basqueira, M.; Selegatto, G.; Cervato, M.C.; Duarte-Neto, A.N.; Higashino, H.R.; et al. Sabiá Virus-Like Mammarenavirus in Patient with Fatal Hemorrhagic Fever, Brazil, 2020. Emerg. Infect. Dis. 2020, 26, 1332–1334. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Pullan, S.T.; Lemey, P.; Suchard, M.A.; Ehichioya, D.U.; Pahlmann, M.; Thielebein, A.; Hinzmann, J.; Oestereich, L.; Wozniak, D.M.; et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 2019, 363, 74–77. [Google Scholar] [CrossRef]
- Zhao, L.; Shi, Y.; Lau, H.C.; Liu, W.; Luo, G.; Wang, G.; Liu, C.; Pan, Y.; Zhou, Q.; Ding, Y.; et al. Uncovering 1058 Novel Human Enteric DNA Viruses Through Deep Long-Read Third-Generation Sequencing and Their Clinical Impact. Gastroenterology 2022, 163, 699–711. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Hoots, W.K. History of plasma-product safety. Transfus. Med. Rev. 2001, 15, 3–10. [Google Scholar] [CrossRef]
- López-Menchero, C.; Alvarez, M.; Fernández, P.; Guzmán, M.; Ortiz-de-Salazar, M.I.; Arbona, C. Evolution of the residual risk of HBV, HCV and HIV transmission through blood transfusion in the Region of Valencia, Spain, during a 15-year period (2003–2017). Blood Transfus. 2019, 17, 418–427. [Google Scholar] [CrossRef]
- Ouattara, H.; Siransy-Bogui, L.; Fretz, C.; Diane, K.M.; Konate, S.; Koidio, A.; Minga, K.A.; Hyda, J.; Koffi-Abe, N.; Offoumou, A.M.; et al. Residual risk of HIV, HVB and HCV transmission by blood transfusion between 2002 and 2004 at the Abidjan National Blood Transfusion Center. Transfus. Clin. Biol. 2006, 13, 242–245. [Google Scholar] [CrossRef]
- Vermeulen, M.; Lelie, N.; Coleman, C.; Sykes, W.; Jacobs, G.; Swanevelder, R.; Busch, M.; van Zyl, G.; Grebe, E.; Welte, A.; et al. Assessment of HIV transfusion transmission risk in South Africa: A 10-year analysis following implementation of individual donation nucleic acid amplification technology testing and donor demographics eligibility changes. Transfusion 2019, 59, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Slavov, S.N.; Santos, E.V.; Hespanhol, M.R.; Rodrigues, E.S.; Haddad, R.; Ubiali, E.M.A.; Covas, D.T.; Kashima, S. Dengue RNA detection and seroprevalence in blood donors during an outbreak in the São Paulo State, Brazil, 2016. J. Med. Virol. 2021, 93, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Custer, B.; Grebe, E.; Buccheri, R.; Bakkour, S.; Stone, M.; Capuani, L.; Alencar, C.; Amorim, L.; Loureiro, P.; Carneiro-Proietti, A.B.; et al. Surveillance for Zika, chikungunya and dengue virus incidence and RNAemia in blood donors at four Brazilian blood centers during 2016–2019. J. Infect. Dis. 2022, jiac173. [Google Scholar] [CrossRef] [PubMed]
- Levi, J.E.; Nishiya, A.; Félix, A.C.; Salles, N.A.; Sampaio, L.R.; Hangai, F.; Sabino, E.C.; Mendrone, A., Jr. Real-time symptomatic case of transfusion-transmitted dengue. Transfusion 2015, 55, 961–964. [Google Scholar] [CrossRef]
- Sabino, E.C.; Loureiro, P.; Lopes, M.E.; Capuani, L.; McClure, C.; Chowdhury, D.; Di-Lorenzo-Oliveira, C.; Oliveira, L.C.; Linnen, J.M.; Lee, T.H.; et al. Transfusion-Transmitted Dengue and Associated Clinical Symptoms During the 2012 Epidemic in Brazil. J. Infect. Dis. 2016, 213, 694–702. [Google Scholar] [CrossRef]
- Santos, F.L.S.; Slavov, S.N.; Bezerra, R.S.; Santos, E.V.; Silva-Pinto, A.C.; Morais, A.L.L.; Sá, M.B.; Ubiali, E.M.A.; De Santis, G.C.; Covas, D.T.; et al. Vaso-occlusive crisis in a sickle cell patient after transfusion-transmitted dengue infection. Transfusion 2020, 60, 2139–2143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.C.C.; Sridhar, S.; Wong, S.C.; Wong, S.C.Y.; Chan, J.F.W.; Yip, C.C.Y.; Chau, C.H.; Au, T.W.K.; Hwang, Y.Y.; Yau, C.S.W. Japanese Encephalitis Virus Transmitted Via Blood Transfusion, Hong Kong, China. Emerg. Infect. Dis. 2018, 24, 49–57. [Google Scholar] [CrossRef]
- Hoad, V.C.; Speers, D.J.; Keller, A.J.; Dowse, G.K.; Seed, C.R.; Lindsay, M.D.; Faddy, H.M.; Pink, J. First reported case of transfusion-transmitted Ross River virus infection. Med. J. Aust. 2015, 202, 267–270. [Google Scholar] [CrossRef]
- Motta, I.J.; Spencer, B.R.; Cordeiro da Silva, S.G.; Arruda, M.B.; Dobbin, J.A.; Gonzaga, Y.B.; Arcuri, I.P.; Tavares, R.C.; Atta, E.H.; Fernandes, R.F.; et al. Evidence for Transmission of Zika Virus by Platelet Transfusion. N. Engl. J. Med. 2016, 375, 1101–1103. [Google Scholar] [CrossRef]
- Sauvage, V.; Gomez, J.; Boizeau, L.; Laperche, S. The potential of viral metagenomics in blood transfusion safety. Transfus. Clin. Biol. 2017, 24, 218–222. [Google Scholar] [CrossRef]
- Dodd, R.Y.; Leiby, D.A. Emerging infectious threats to the blood supply. Annu. Rev. Med. 2004, 55, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Busch, M.P. Transfusion-transmitted arboviruses. Vox Sang. 2010, 98, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Cebriá-Mendoza, M.; Bracho, M.A.; Arbona, C.; Larrea, L.; Díaz, W.; Sanjuán, R.; Cuevas, J.M. Exploring the Diversity of the Human Blood Virome. Viruses 2021, 13, 2322. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Dzakah, E.E.; Rashid, F.; Wang, J.; Tang, S. The Second Human Pegivirus, a Non-Pathogenic RNA Virus with Low Prevalence and Minimal Genetic Diversity. Viruses 2022, 14, 1844. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel-Abramowski, S.; Taleb, S.; Alessandrini, M.; Preynat-Seauve, O. Viral Metagenomics of Blood Donors and Blood-Derived Products Using Next-Generation Sequencing. Transfus. Med. Hemother. 2019, 46, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhou, Z.; Yao, L.; Xu, Y.; Wang, L.; Fan, X. Full annotation of serum virome in Chinese blood donors with elevated alanine aminotransferase levels. Transfusion 2019, 59, 3177–3185. [Google Scholar] [CrossRef]
- Cebriá-Mendoza, M.; Arbona, C.; Larrea, L.; Díaz, W.; Arnau, V.; Peña, C.; Bou, J.V.; Sanjuán, R.; Cuevas, J.M. Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans. Sci. Rep. 2021, 11, 6921. [Google Scholar] [CrossRef]
- Hsiao, K.L.; Wang, L.Y.; Cheng, J.C.; Cheng, Y.J.; Lin, C.L.; Liu, H.F. Detection and genetic characterization of the novel torque teno virus group 6 in Taiwanese general population. R. Soc. Open Sci. 2021, 8, 210938. [Google Scholar] [CrossRef]
- Bal, A.; Oriol, G.; Josset, L.; Generenaz, L.; Sarkozy, C.; Sesques, P.; Salles, G.; Morfin, F.; Lina, B.; Becker, J.; et al. Metagenomic Investigation of Torque Teno Mini Virus-SH in Hematological Patients. Front. Microbiol. 2019, 10, 1898. [Google Scholar] [CrossRef]
- Allain, J.P.; Thomas, I.; Sauleda, S. Nucleic acid testing for emerging viral infections. Transfus. Med. 2002, 12, 275–283. [Google Scholar] [CrossRef]
- Thijssen, M.; Tacke, F.; Beller, L.; Deboutte, W.; Yinda, K.C.; Nevens, F.; Laleman, W.; Van Ranst, M.; Pourkarim, M.R. Clinical relevance of plasma virome dynamics in liver transplant recipients. EBioMedicine 2020, 60, 103009. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, A.L.; Wunderink, H.F.; Sidorov, I.A.; de Brouwer, C.S.; Kroes, A.C.; Putter, H.; de Vries, A.P.; Rotmans, J.I.; Feltkamp, M.C. Torque teno virus loads after kidney transplantation predict allograft rejection but not viral infection. J. Clin. Virol. 2021, 140, 104871. [Google Scholar] [CrossRef] [PubMed]
- Giménez, E.; Monzó, C.; Albert, E.; Fuentes-Trillo, A.; Seda, E.; Piñana, J.L.; Hernández Boluda, J.C.; Solano, C.; Chaves, J.; Navarro, D. Diversity and dynamic changes of anelloviruses in plasma following allogeneic hematopoietic stem cell transplantation. J. Med. Virol. 2021, 93, 5167–5172. [Google Scholar] [CrossRef] [PubMed]
- Mouton, W.; Conrad, A.; Bal, A.; Boccard, M.; Malcus, C.; Ducastelle-Lepretre, S.; Balsat, M.; Barraco, F.; Larcher, M.V.; Fossard, G.; et al. Torque Teno Virus Viral Load as a Marker of Immune Function in Allogeneic Haematopoietic Stem Cell Transplantation Recipients. Viruses 2020, 12, 1292. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Dai, R.; Zhang, X. Global prevalence of human pegivirus-1 in healthy volunteer blood donors: A systematic review and meta-analysis. Vox Sang. 2020, 115, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, S.; Schmidt, R.; Day, T. Fighting the Public Health Burden of AIDS With the Human Pegivirus. Am. J. Epidemiol. 2019, 188, 1586–1594. [Google Scholar] [CrossRef]
- Samadi, M.; Salimi, V.; Haghshenas, M.R.; Miri, S.M.; Mohebbi, S.R.; Ghaemi, A. Clinical and molecular aspects of human pegiviruses in the interaction host and infectious agent. Virol. J. 2022, 19, 41. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Deng, X.; Blümel, J.; Nübling, C.M.; Hunfeld, A.; Baylis, S.A.; Delwart, E. Viral nucleic acids in human plasma pools. Transfusion 2016, 56, 2248–2255. [Google Scholar] [CrossRef]
- Dos Santos Bezerra, R.; Bitencourt, H.T.; Covas, D.T.; Kashima, S.; Slavov, S.N. Molecular evolution pattern of Merkel cell polyomavirus identified by viral metagenomics in plasma of high-risk blood donors from the Brazilian Amazon. Infect. Genet. Evol. 2020, 85, 104563. [Google Scholar] [CrossRef]
- Bezerra, R.D.S.; Ximenez, J.P.B.; Giovanetti, M.; Zucherato, V.S.; Bitencourt, H.T.; Zimmermann, A.; Alcantara, L.C.J.; Covas, D.T.; Kashima, S.; Slavov, S.N. Metavirome composition of Brazilian blood donors positive for the routinely tested blood-borne infections. Virus Res. 2022, 311, 198689. [Google Scholar] [CrossRef]
- Dos Santos Bezerra, R.; de Oliveira, L.S.; Moretto, E.L.; Amorim Ubiali, E.M.; Silveira, R.M.; da Silva, W.A. Junior.; Covas, D.T.; Kashima, S.; Slavov, S.N. Viral metagenomics in blood donations with post-donation illness reports from Brazil. Blood Transfus. 2021, 19, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Bezerra, R.; de Melo Jorge, D.M.; Castro, Í.A.; Moretto, E.L.; Scalon de Oliveira, L.; Ubiali, E.M.A.; Covas, D.T.; Arruda, E.; Kashima, S.; Slavov, S.N. Detection of Influenza A(H3N2) Virus RNA in Donated Blood. Emerg. Infect. Dis. 2020, 26, 1621–1623. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef]
- Wanzeller, A.L.; Souza, A.L.; Azevedo, R.S.; Júnior, E.C.; Filho, L.C.; Oliveira, R.S.; Lemos, P.S.; Júnior, J.V.; Vasconcelos, P.F. Complete Genome Sequence of the BeAn 58058 Virus Isolated from Oryzomys sp. Rodents in the Amazon Region of Brazil. Genome Announc. 2017, 5, e01575-16. [Google Scholar] [CrossRef]
- Jurasz, H.; Pawłowski, T.; Perlejewski, K. Contamination Issue in Viral Metagenomics: Problems, Solutions, and Clinical Perspectives. Front. Microbiol. 2021, 12, 745076. [Google Scholar] [CrossRef]
- Simner, P.J.; Salzberg, S.L. The Human “Contaminome” and Understanding Infectious Disease. N. Engl. J. Med. 2022, 387, 943–946. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Ounit, R.; Afshinnekoo, E.; Prill, R.J.; Hénaff, E.; Alexander, N.; Minot, S.S.; Danko, D.; Foox, J.; Ahsanuddin, S.; et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017, 18, 182. [Google Scholar] [CrossRef]
- Gräf, T.; Vazquez, C.; Giovanetti, M.; de Bruycker-Nogueira, F.; Fonseca, V.; Claro, I.M.; de Jesus, J.G.; Gómez, A.; Xavier, J.; de Mendonça, M.C.L.; et al. Epidemiologic History and Genetic Diversity Origins of Chikungunya and Dengue Viruses, Paraguay. Emerg. Infect. Dis. 2021, 27, 1393–1404. [Google Scholar] [CrossRef]
- Giovanetti, M.; Pereira, L.A.; Adelino, T.É.R.; Fonseca, V.; Xavier, J.; de Araújo Fabri, A.; Slavov, S.N.; da Silva Lemos, P.; de Almeida Marques, W.; Kashima, S.; et al. A Retrospective Overview of Zika Virus Evolution in the Midwest of Brazil. Microbiol. Spectr. 2022, 10, e0015522. [Google Scholar] [CrossRef]
- Sauvage, V.; Boizeau, L.; Candotti, D.; Vandenbogaert, M.; Servant-Delmas, A.; Caro, V.; Laperche, S. Early MinION™ nanopore single-molecule sequencing technology enables the characterization of hepatitis B virus genetic complexity in clinical samples. PLoS ONE 2018, 13, e0194366. [Google Scholar] [CrossRef] [PubMed]
- Saá, P.; Fink, R.V.; Bakkour, S.; Jin, J.; Simmons, G.; Muench, M.O.; Dawar, H.; Di Germanio, C.; Hui, A.J.; Wright, D.J.; et al. Frequent detection but lack of infectivity of SARS-CoV-2 RNA in pre-symptomatic, infected blood donor plasma. J. Clin. Investig. 2022, e159876. [Google Scholar] [CrossRef] [PubMed]
- Cappy, P.; Candotti, D.; Sauvage, V.; Lucas, Q.; Boizeau, L.; Gomez, J.; Enouf, V.; Chabli, L.; Pillonel, J.; Tiberghien, P.; et al. No evidence of SARS-CoV-2 transfusion transmission despite RNA detection in blood donors showing symptoms after donation. Blood 2020, 136, 1888–1891. [Google Scholar] [CrossRef] [PubMed]
- Stramer, S.L.; Hollinger, F.B.; Katz, L.M.; Kleinman, S.; Metzel, P.S.; Gregory, K.R.; Dodd, R.Y. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009, 49, 1S–29S. [Google Scholar] [CrossRef] [PubMed]
- Dean, C.L.; Wade, J.; Roback, J.D. Transfusion-Transmitted Infections: An Update on Product Screening, Diagnostic Techniques, and the Path Ahead. J. Clin. Microbiol. 2018, 56, e00352-18. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, D.; Matsvay, A.; Abramov, I.; Dedkov, V.; Shipulin, G.; Khafizov, K. Current Trends in Diagnostics of Viral Infections of Unknown Etiology. Viruses 2020, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.P.; Nuebling, C.M.; Lery, F.X.; Maryuningsih, Y.S.; Epstein, J.S. A WHO tool for risk-based decision making on blood safety interventions. Transfusion 2021, 61, 503–515. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slavov, S.N. Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine. Viruses 2022, 14, 2448. https://doi.org/10.3390/v14112448
Slavov SN. Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine. Viruses. 2022; 14(11):2448. https://doi.org/10.3390/v14112448
Chicago/Turabian StyleSlavov, Svetoslav Nanev. 2022. "Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine" Viruses 14, no. 11: 2448. https://doi.org/10.3390/v14112448
APA StyleSlavov, S. N. (2022). Viral Metagenomics for Identification of Emerging Viruses in Transfusion Medicine. Viruses, 14(11), 2448. https://doi.org/10.3390/v14112448