
Host Cell Redox Alterations Promote Latent HIV-1 Reactivation through Atypical Transcription Factor Cooperativity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. A Small Compound Screen Identifies a Novel Class of HIV-1 Latency-Reversing Agents
2.2. SMOREs Induce HIV-1 RNA Processing and Function Combinatorially with Epigenetic LRAs
2.3. Transcriptome Profiling Unveils Induction of Host Cell Master Transcriptional Regulators in Response to Oxidative Stress Cell Signaling
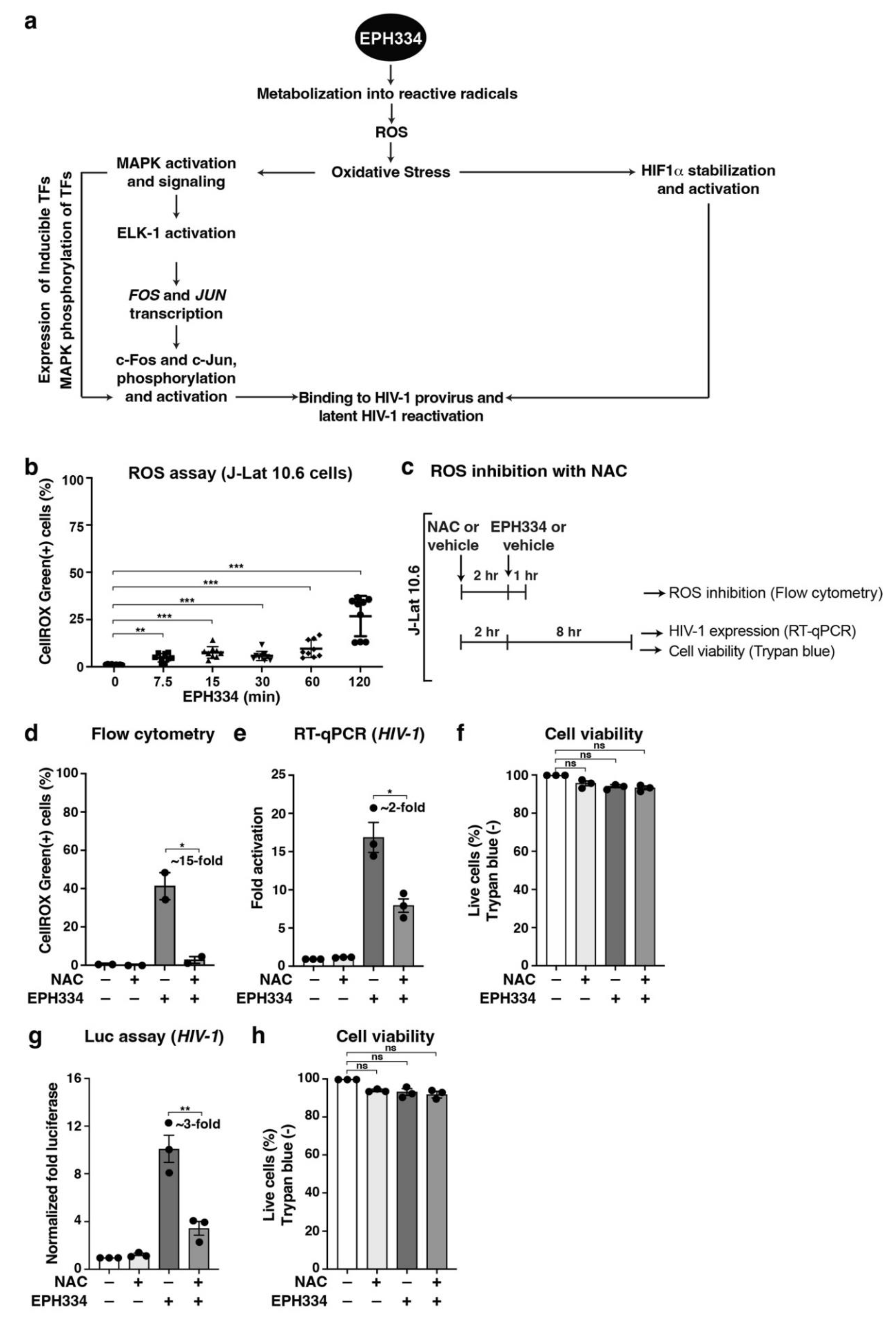
2.4. EPH334 Induces Oxidative Stress to Stimulate Latent HIV-1 Reactivation in CD4+ T Cell Models of Latency
2.5. EPH334 Stimulates Redox-Sensitive TFs That Mediate Latent HIV-1 Reactivation
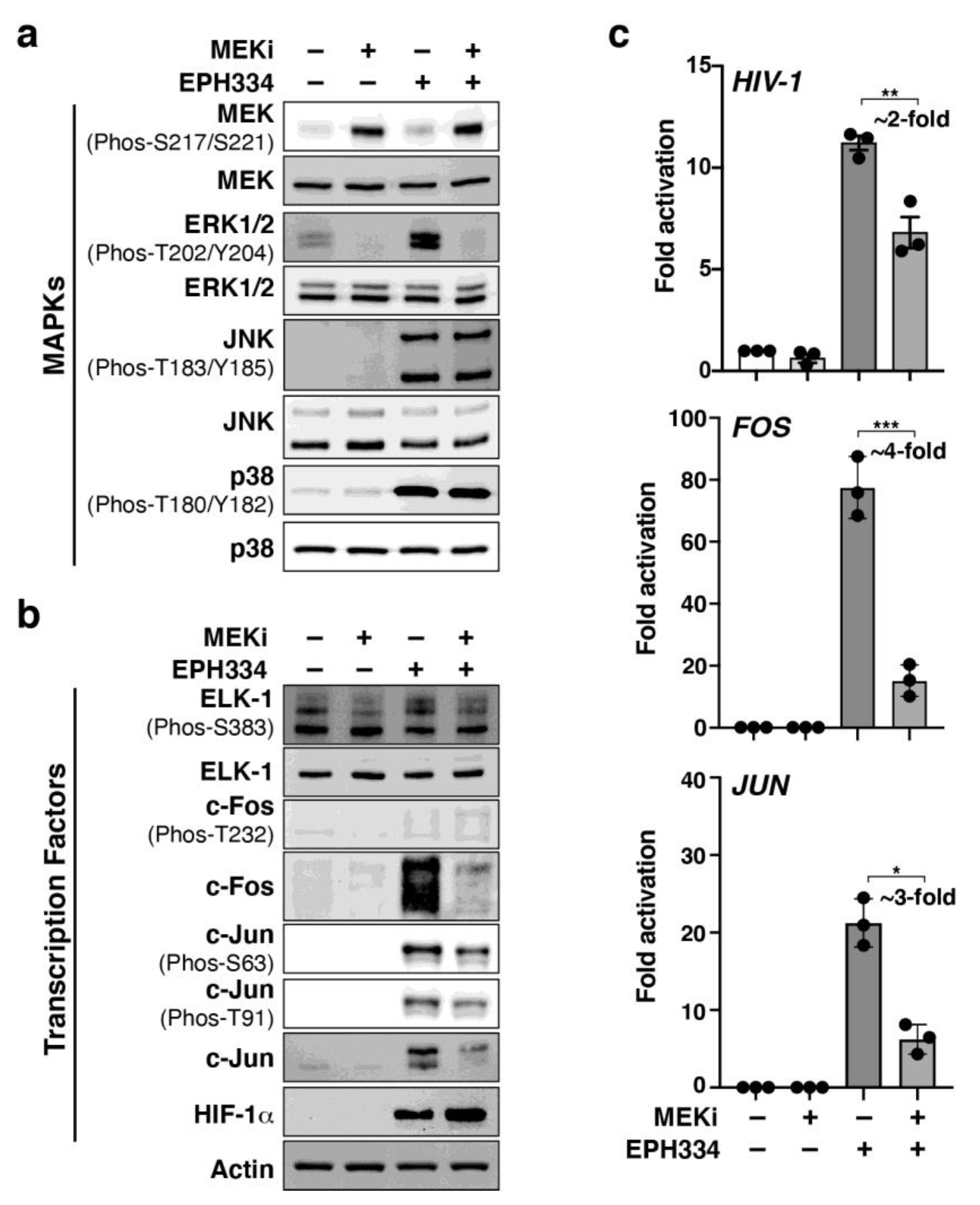
2.6. EPH334 Induces Redox-Sensitive Signaling Kinases and TFs That Reactivate Latent HIV-1 in a ROS-Dependent Manner
2.7. A Redox-Sensitive MEK–ERK1/2 Axis Facilitates EPH334-Induced Latent HIV-1 Reactivation
3. Discussion
4. Methods
4.1. SMOREs and EPH334
4.2. Cell Culture
4.3. Nucleofection of Jurkat Cells and RNAi
4.4. Flow Cytometry Analysis
4.5. RNA Extraction and RT-qPCR Assays
4.6. RNA-seq
4.7. RNA-seq Data Analysis
4.8. Oxidative Stress Detection
4.9. ChIP-qPCR Assays
4.10. Isolation of Naïve CD4+ T Cells and Generation of TCM
4.11. Primary Model of Latency
4.12. Generation of Polarized Effector CD4+ T Cells
4.13. Isolation of Resting CD4+ T Cells from Patient Samples and REVEAL Assay
4.14. Data Deposition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.-G.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell. Rep. 2019, 27, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim. Biophys. Acta 2007, 1773, 1285–1298. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Kwon, H.; Chen, L.F.; Greene, W.C. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 2007, 81, 6043–6056. [Google Scholar] [CrossRef] [PubMed]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Ellitson, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef]
- Duette, G.; Pereyra Gerber, P.; Rubione, J.; Perez, P.S.; Landay, A.L.; Crowe, S.M.; Liao, Z.; Witwer, K.W.; Holgado., M.P.; Salido, J.; et al. Induction of HIF-1alpha by HIV-1 Infection in CD4(+) T Cells Promotes Viral Replication and Drives Extracellular Vesicle-Mediated Inflammation. mBio 2018, 9, 3607–3612. [Google Scholar] [CrossRef]
- Das, B.; Dobrowolski, C.; Luttge, B.; Valadkhan, S.; Chomont, N.; Jonston, R.; Bacchetti, P.; Hoh, R.; Gandhi, M.; Deeks, S.G.; et al. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proc. Natl. Acad. Sci. USA 2018, 115, E7795–E7804. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Ramirez, N.P.; D’Orso, I. HIV-1 Proviral Transcription and Latency in the New Era. Viruses 2020, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef]
- Fong, L.E.; Sulistijo, E.S.; Miller-Jensen, K. Systems analysis of latent HIV reversal reveals altered stress kinase signaling and increased cell death in infected T cells. Sci. Rep. 2017, 7, 16179. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.D.; Dull, A.B.; Beutler, J.A.; Hager, G.L. High-content fluorescence-based screening for epigenetic modulators. Methods Enzymol. 2006, 414, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Hojfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef]
- Easmon, J.; Heinisch, G.; Purstinger, G.; Langer, T.; Österreicher, J.K.; Grunicke, H.H.; Hofmann, J. Azinyl and diazinyl hydrazones derived from aryl N-heteroaryl ketones: Synthesis and antiproliferative activity. J. Med. Chem. 1997, 40, 4420–4425. [Google Scholar] [CrossRef]
- Easmon, J.; Pürstinger, G.; Heinisch, G.; Fiebig, H.H.; Roth, T.; Hofmann, J. Synthesis, cytotoxic, and antitumor activities of 2-pyridylhydrazones derived from 3-benzoylpyridazines. Arch. Pharm. 2014, 347, 599–608. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.D.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef]
- Khan, S.Z.; Hand, N.; Zeichner, S.L. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4(+) T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Alireza Rabi, S.; Laird, G.M.; Eisele, E.E.; Zhang, H.; Margolick, J.B.; Siliciano, R.F. A novel PCR assay for quantification of HIV-1 RNA. J. Virol. 2013, 87, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2019, 10, 3060. [Google Scholar] [CrossRef]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef]
- Reeder, J.E.; Kwak, Y.T.; McNamara, R.P.; Forst, C.V.; D’Orso, I. HIV Tat controls RNA Polymerase II and the epigenetic landscape to transcriptionally reprogram target immune cells. Elife 2015, 4, e08955. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnick, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Pope, S.D.; Medzhitov, R. Emerging Principles of Gene Expression Programs and Their Regulation. Mol. Cell 2018, 71, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 2011, 16, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxid. Med. Cell Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [PubMed]
- Himanen, S.V.; Sistonen, L. New insights into transcriptional reprogramming during cellular stress. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Su, C.; Gao, G.; Schneider, S.; Helt, C.; Weiss, C.; O’Reilly, M.A.; Bohmann, D.; Zhao, J. DNA damage induces downregulation of histone gene expression through the G1 checkpoint pathway. EMBO J. 2004, 23, 1133–1143. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Gu, D.S.; Kagnoff, M.F. Activating protein-1 cooperates with phorbol ester activation signals to increase HIV-1 expression. AIDS 1996, 10, 819–826. [Google Scholar] [CrossRef]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Mukerjee, R.; Fan, S.; Del Valle, L.; Michiels, C.; Sweet, T.; Rom, I.; Khalili, K.; Rapaport, J.; Amini, S.; et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J. Biol. Chem. 2009, 284, 11364–11373. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Messi, M.; Giacchetto, I.; Nagata, K.; Lanzavecchia, A.; Natoli, G.; Sallusto, F. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat. Immunol. 2003, 4, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, I.; Novis, C.L.; Macedo, A.B.; Takata, H.; Nell, R.; Kakazu, J.C.; Furler, R.L.; Shakya, B.; Schubert, H.L.; Hill, C.P.; et al. Activation of the Anti-Oxidative Stress Response Reactivates Latent HIV-1 Through the Mitochondrial Antiviral Signaling Protein Isoform MiniMAVS. Front. Immunol. 2021, 12, 682182. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Shumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Sun, H.; Kim, D.; Li, X.; Kiselinova, M.; Ouyang, Z.; Vandekerchove, L.; Shang, H.; Rosenberg, E.S.; Yu, X.G.; Lichterfeld, M. Th1/17 Polarization of CD4 T Cells Supports HIV-1 Persistence during Antiretroviral Therapy. J. Virol. 2015, 89, 11284–11293. [Google Scholar] [CrossRef]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.-H.; Hua, S.; Chen, H.-R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 2017, 127, 2689–2696. [Google Scholar] [CrossRef]
- Alfranca, A.; Dolores Gutierrez, M.; Vara, A.; Aragones, J.; Vidal, F.; Landázuri, M.O. c-Jun and hypoxia-inducible factor 1 functionally cooperate in hypoxia-induced gene transcription. Mol. Cell Biol. 2002, 22, 12–22. [Google Scholar] [CrossRef]
- Bannister, A.J.; Brown, H.J.; Sutherland, J.A.; Kouzarides, T. Phosphorylation of the c-Fos and c-Jun HOB1 motif stimulates its activation capacity. Nucleic Acids Res. 1994, 22, 5173–5176. [Google Scholar] [CrossRef]
- Babu, G.J.; Lalli, M.J.; Sussman, M.A.; Sadoshima, J.; Periasamy, M. Phosphorylation of elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. J. Mol. Cell Cardiol. 2000, 32, 1447–1457. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Hunter, T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr. Biol. 1995, 5, 747–757. [Google Scholar] [CrossRef]
- Yang, S.H.; Yates, P.R.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. The Elk-1 ETS-domain transcription factor contains a mitogen-activated protein kinase targeting motif. Mol. Cell Biol. 1998, 18, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Cruzalegui, F.H.; Cano, E.; Treisman, R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 1999, 18, 7948–7957. [Google Scholar] [CrossRef]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Gassmann, M.; Marti, H.H. Hypoxia-inducible factor-1 alpha is regulated at the post-mRNA level. Kidney Int. 1997, 51, 560–563. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Staal, F.J.; Roederer, M.; Herzenberg, L.A.; Herzenberg, L.A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1990, 87, 9943–9947. [Google Scholar] [CrossRef]
- Pyo, C.W.; Yang, Y.L.; Yoo, N.K.; Choi, S.Y. Reactive oxygen species activate HIV long terminal repeat via post-translational control of NF-kappaB. Biochem. Biophys. Res. Commun. 2008, 376, 180–185. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [PubMed]
- Schiralli Lester, G.M.; Henderson, A.J. Mechanisms of HIV Transcriptional Regulation and Their Contribution to Latency. Mol. Biol. Int. 2012, 2012, 614120. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Amini, S.; Sen, S.; Khalili, K.; Sawaya, B.E. Regulation of the HIV-1 promoter by HIF-1alpha and Vpr proteins. Virol. J. 2011, 8, 477. [Google Scholar] [CrossRef] [PubMed]
- Damert, A.; Ikeda, E.; Risau, W. Activator-protein-1 binding potentiates the hypoxia-induciblefactor-1-mediated hypoxia-induced transcriptional activation of vascular-endothelial growth factor expression in C6 glioma cells. Biochem. J. 1997, 327 Pt 2, 419–423. [Google Scholar] [CrossRef]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates III, J.R. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1 Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef]
- Mbonye, U.; Leskov, K.; Shukla, M.; Valadkhan, S.; Karn, J. Biogenesis of P-TEFb in CD4+ T cells to reverse HIV latency is mediated by protein kinase C (PKC)-independent signaling pathways. PLoS Pathog. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- McNamara, R.P.; Bacon, C.W.; D’Orso, I. Transcription Elongation Control by the 7SK snRNP Complex: Releasing the Pause. Cell Cycle 2016, 15, 2115–2123. [Google Scholar] [CrossRef]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2018, 10, 1–19. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, T.; Shang, C.; Luo, Y.; Liu, L.; Yan, J.; Li, Y.; Huang, S. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget 2014, 5, 10140–10150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, J.; Zhang, L.; Wang, M.; Zhou, L.; Feng, X.; Yu, L.; Lan, J.; Gao, W.; Zhang, C.; Bu, Y.; et al. CPX Targeting DJ-1 Triggers ROS-induced Cell Death and Protective Autophagy in Colorectal Cancer. Theranostics 2019, 9, 5577–5594. [Google Scholar] [CrossRef] [PubMed]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoeck, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [CrossRef]
- Price, M.A.; Cruzalegui, F.H.; Treisman, R. The p38 and ERK MAP kinase pathways cooperate to activate Ternary Complex Factors and c-fos transcription in response to UV light. EMBO J. 1996, 15, 6552–6563. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 1–10. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McNamara, R.P.; Reeder, J.E.; McMillan, E.A.; Bacon, C.W.; McCann, J.L.; D’Orso, I. KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol. Cell 2016, 61, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, N.P.; Lee, J.; Zheng, Y.; Li, L.; Dennis, B.; Chen, D.; Challa, A.; Planelles, V.; Westover, K.D.; Alto, N.M.; et al. ADAP1 promotes latent HIV-1 reactivation by selectively tuning KRAS-ERK-AP-1 T cell signaling-transcriptional axis. Nat. Commun. 2022, 13, 1109. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Lorenzo, E.; Ramirez, N.-G.P.; Lee, J.; Pandhe, S.; Wang, L.; Hernandez-Doria, J.; Spivak, A.M.; Planelles, V.; Petersen, T.; Jain, M.K.; et al. Host Cell Redox Alterations Promote Latent HIV-1 Reactivation through Atypical Transcription Factor Cooperativity. Viruses 2022, 14, 2288. https://doi.org/10.3390/v14102288
Cruz-Lorenzo E, Ramirez N-GP, Lee J, Pandhe S, Wang L, Hernandez-Doria J, Spivak AM, Planelles V, Petersen T, Jain MK, et al. Host Cell Redox Alterations Promote Latent HIV-1 Reactivation through Atypical Transcription Factor Cooperativity. Viruses. 2022; 14(10):2288. https://doi.org/10.3390/v14102288
Chicago/Turabian StyleCruz-Lorenzo, Emily, Nora-Guadalupe P. Ramirez, Jeon Lee, Sonali Pandhe, Lei Wang, Juan Hernandez-Doria, Adam M. Spivak, Vicente Planelles, Tianna Petersen, Mamta K. Jain, and et al. 2022. "Host Cell Redox Alterations Promote Latent HIV-1 Reactivation through Atypical Transcription Factor Cooperativity" Viruses 14, no. 10: 2288. https://doi.org/10.3390/v14102288
APA StyleCruz-Lorenzo, E., Ramirez, N.-G. P., Lee, J., Pandhe, S., Wang, L., Hernandez-Doria, J., Spivak, A. M., Planelles, V., Petersen, T., Jain, M. K., Martinez, E. D., & D’Orso, I. (2022). Host Cell Redox Alterations Promote Latent HIV-1 Reactivation through Atypical Transcription Factor Cooperativity. Viruses, 14(10), 2288. https://doi.org/10.3390/v14102288