


Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Creation of Test Data
2.2. Creation of Custom Viral Sequence Databases
2.3. Creation of Custom Viral HMM Databases
2.4. Testing of Sequence-Based Classification Tools
2.5. Construction of Mock Libraries for Workflow Evaluation
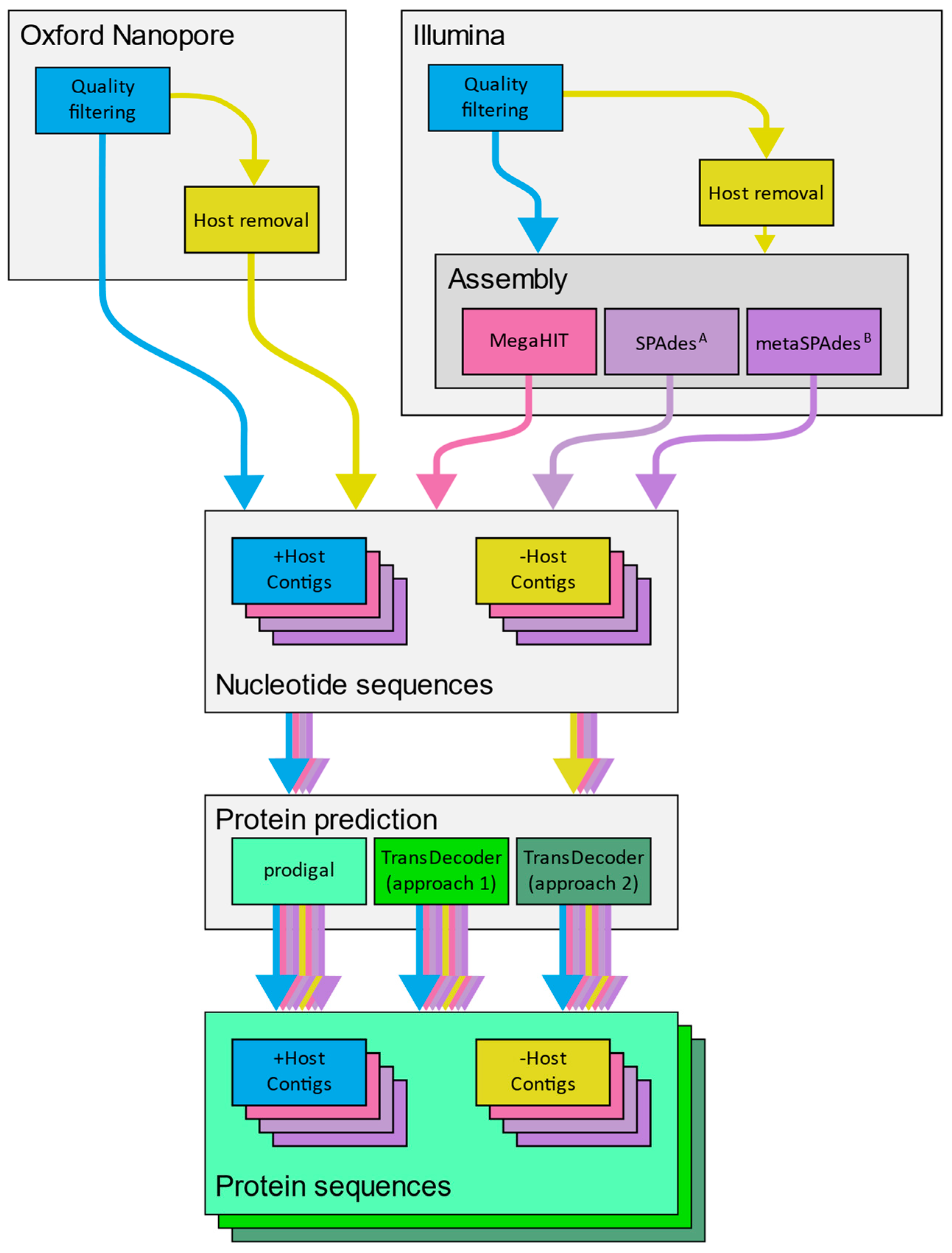
2.6. Application of a Diagnostic Workflows to Mock Libraries
3. Results and Discussion
3.1. Creation of Target and Non-Target Datasets for Tool Testing
3.2. Creation of Custom Sequence Databases
3.3. Creation and Assessment of Custom Viral HMM Databases
3.4. Classification and Database Profiling
3.5. Recovery of Nucleotide and Gene Sequences from Simulated Libraries
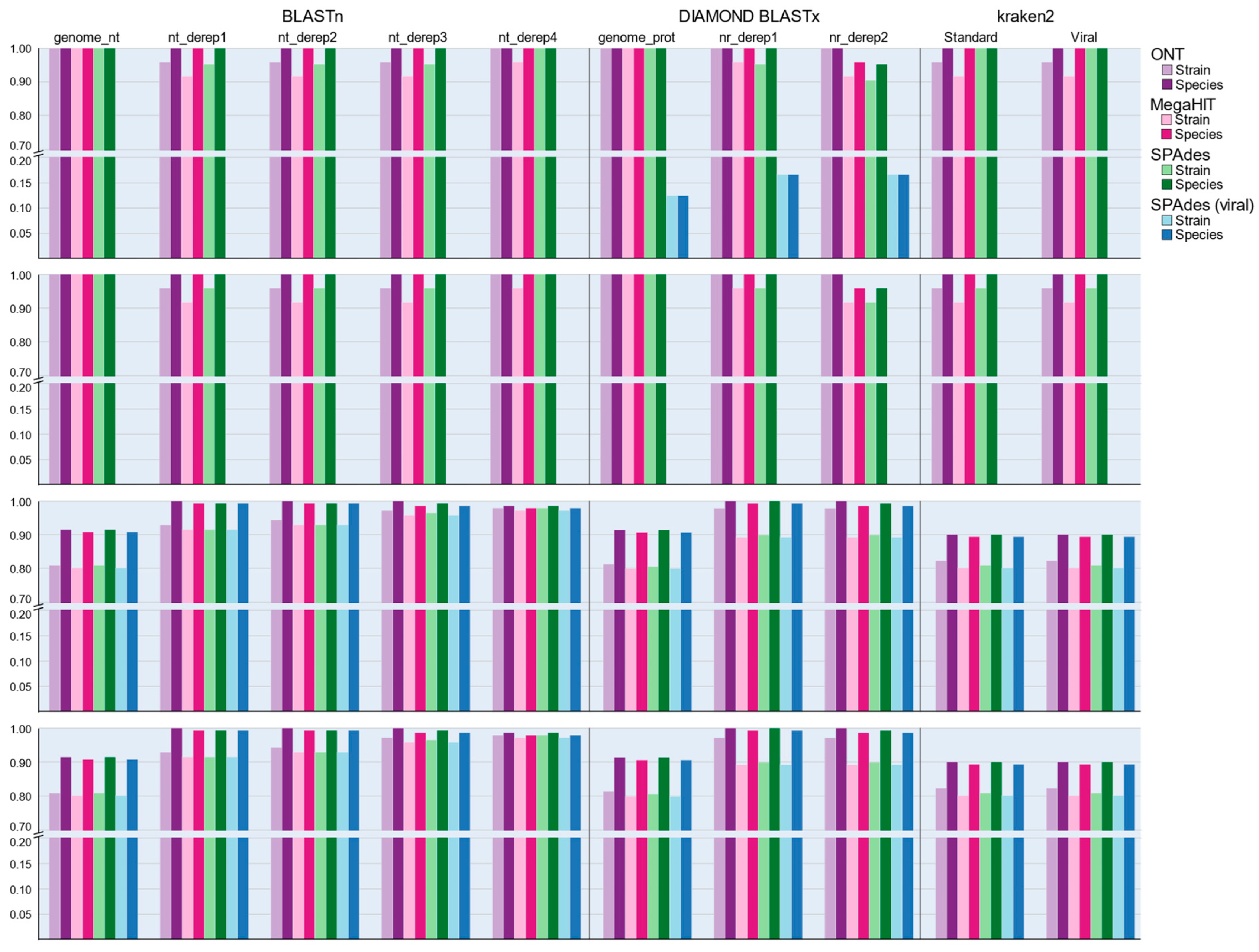
3.6. Performance of Nucleotide-Based Classification Approaches
3.7. Performance of Protein–Protein Classification Approaches
3.8. Performance of Analysis Workflows against VIROMOCK Data
3.9. Challenges in Creating a Stand-Alone Analysis Pipeline
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- An Overview of New Zealand’s Trade in 2021. Available online: https://www.mfat.govt.nz/assets/Trade/MFAT-Market-reports/An-overview-of-New-Zealands-trade-in-2021-March-2021.pdf (accessed on 3 June 2022).
- Situation and Outlook for Primary Industries (SOPI) Data. Available online: https://www.mpi.govt.nz/resources-and-forms/economic-intelligence/data/ (accessed on 1 July 2022).
- Stone, M. Biosecurity is critical to New Zealand’s national security, economy and way of life. N. Z. Vet. J. 2021, 69, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Souiri, A.; Khataby, K.; Kasmi, Y.; Zemzami, M.; Amzazi, S.; Ennaji, M.M. Chapter 16—Risk Assessment and Biosecurity Considerations in Control of Emergent Plant Viruses. In Emerging and Reemerging Viral Pathogens; Academic Press: Cambridge, MA, USA, 2020; pp. 287–311. [Google Scholar]
- Foot-and-Mouth Disease Economic Impact Assessment: What it means for New Zealand. Available online: https://www.mpi.govt.nz/dmsdocument/4406-Foot-and-Mouth-Disease-Economic-Impact-Assessment-What-it-means-for-New-Zealand (accessed on 1 July 2022).
- Tuppurainen, E.S.M.; Venter, E.H.; Shisler, J.L.; Gari, G.; Mekonnen, G.A.; Juleff, N.; Lyons, N.A.; De Clercq, K.; Upton, C.; Bowden, T.R.; et al. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound. Emerg. Dis. 2015, 64, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.R.; Miller, R.S.; McKee, S.C.; Ernst, K.H.; Didero, N.M.; Maison, R.M.; Grady, M.J.; Shwiff, S.A. Risks of intro-duction and economic consequences associated with African swine fever, classical swine fever and foot-and-mouth disease: A review of the literature. Transbound. Emerg. Dis. 2021, 68, 1910–1965. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.; Hall, W.; Neumann, E. African swine fever: A New Zealand perspective on epidemiological risk factors for its occurrence. N. Z. Vet. J. 2021, 69, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Ma, T.; Hao, M.; Ding, F.; Sun, K.; Wang, Q.; Kang, T.; Di Wang, D.; Zhao, S.; Li, M.; et al. Quantifying risk factors and potential geographic extent of African swine fever across the world. PLoS ONE 2022, 17, e0267128. [Google Scholar] [CrossRef] [PubMed]
- Keeping African Swine Fever Out of Australia. Available online: https://www.agriculture.gov.au/biosecurity-trade/pests-diseases-weeds/animal/asf (accessed on 1 July 2022).
- Technical Advice: Risk of Lumpy Skin Disease Via Import of Cattle and Buffalo Meat and Meat Products for Human and Ani-mal Consumption. Available online: https://www.mpi.govt.nz/dmsdocument/51352-Technical-Advice-Risk-of-lumpy-skin-disease-via-import-of-cattle-and-buffalo-meat-and-meat-products-for-human-and-animal-consumption (accessed on 1 July 2022).
- Liefting, L.; Waite, D.; Thompson, J. Application of Oxford Nanopore Technology to Plant Virus Detection. Viruses 2021, 13, 1424. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Mumford, R.A.; Macarthur, R.; Boonham, N. The role and challenges of new diagnostic technology in plant biosecurity. Food Secur. 2015, 8, 103–109. [Google Scholar] [CrossRef]
- Boykin, L.; Ghalab, A.; De Marchi, B.R.; Savill, A.; Wainaina, J.M.; Kinene, T.; Lamb, S.; Rodrigues, M.; Kehoe, M.; Ndunguru, J.; et al. Real time portable genome sequencing for global food security. F1000Research 2018, 7, 1101. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef]
- Hu, Y.; Wilson, S.; Schwessinger, B.; Rathjen, J.P. Blurred lines: Integrating emerging technologies to advance plant biosecurity. Curr. Opin. Plant Biol. 2020, 56, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Mehetre, G.; Leo, V.; Singh, G.; Sorokan, A.; Maksimov, I.; Yadav, M.; Upadhyaya, K.; Hashem, A.; Alsaleh, A.; Dawoud, T.; et al. Current Developments and Challenges in Plant Viral Diagnostics: A Systematic Review. Viruses 2021, 13, 412. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of un-known soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a meta-genome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Candresse, T.; Filloux, D.; Muhire, B.; Julian, C.; Galzi, S.; Fort, G.; Bernardo, P.; Daugrois, J.-H.; Fernandez, E.; Martin, D.P.; et al. Appearances Can Be Deceptive: Revealing a Hidden Viral Infection with Deep Sequencing in a Plant Quarantine Context. PLoS ONE 2014, 9, e102945. [Google Scholar] [CrossRef] [PubMed]
- Badial, A.B.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore Sequencing as a Surveillance Tool for Plant Pathogens in Plant and Insect Tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [PubMed]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 2018, 68, 229–238. [Google Scholar] [CrossRef]
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-based detection and characterization of yam viruses. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Fellers, J.P.; Webb, C.; Fellers, M.C.; Rupp, J.S.; De Wolf, E. Wheat Virus Identification within Infected Tissue Using Nanopore Sequencing Technology. Plant Dis. 2019, 103, 2199–2203. [Google Scholar] [CrossRef]
- Della Bartola, M.; Byrne, S.; Mullins, E. Characterization of Potato Virus Y Isolates and Assessment of Nanopore Sequencing to Detect and Genotype Potato Viruses. Viruses 2020, 12, 478. [Google Scholar] [CrossRef]
- Monger, W.A.; Goodfellow, H.A.; Back, E.J. First report of Arabis mosaic virus in potato (Solanum tuberosum), identified by nanopore sequencing. New Dis. Rep. 2020, 41, 29. [Google Scholar] [CrossRef]
- Ziebell, H.; De Jonghe, K.; Rott, M.; Nicolaisen, M.; Gentit, P.; Renvoise, J.-P.; Candresse, T.; Fox, A.; Varveri, C.; Melika, G.; et al. The application of Next-Generation Sequencing technology for the detection and diagnosis of non-culturable organisms: Viruses and viroids (NGSdetect). Zenodo 2021. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C. Clinical Metagenomic Sequencing for Di-agnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.J.C.; Brown, J.R.; Fischer, N.; Sidorov, I.A.; Morfopoulou, S.; Huang, J.; Munnink, B.B.O.; Sayiner, A.; Bulgurcu, A.; Rodriguez, C.; et al. Benchmark of thirteen bioinformatic pipelines for metagenomic virus diagnostics using datasets from clinical samples. J. Clin. Virol. 2021, 141, 104908. [Google Scholar] [CrossRef]
- Carbo, E.C.; Sidorov, I.A.; van Rijn-Klink, A.L.; Pappas, N.; van Boheemen, S.; Mei, H.; Hiemstra, P.S.; Eagan, T.M.; Claas, E.C.J.; Kroes, A.C.M.; et al. Performance of Five Metagenomic Classifiers for Virus Pathogen Detection Using Respiratory Samples from a Clinical Cohort. Pathogens 2022, 11, 340. [Google Scholar] [CrossRef]
- Ibaba, J.D.; Gubba, A. High-Throughput Sequencing Application in the Diagnosis and Discovery of Plant-Infecting Viruses in Africa, A Decade Later. Plants 2020, 9, 1376. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef]
- Susi, H.; Filloux, D.; Frilander, M.J.; Roumagnac, P.; Laine, A.-L. Diverse and variable virus communities in wild plant populations revealed by metagenomic tools. PeerJ 2019, 7, e6140. [Google Scholar] [CrossRef]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.A.; Lelwala, R.V.; Elliott, C.E.; Windell, C.; Fiorito, S.; Dinsdale, A.; Whattam, M.; Pattemore, J.; Barrero, R.A. Side-by-Side Comparison of Post-Entry Quarantine and High Throughput Sequencing Methods for Virus and Viroid Diagnosis. Biology 2022, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Rampelli, S.; Soverini, M.; Turroni, S.; Quercia, S.; Biagi, E.; Brigidi, P.; Candela, M. ViromeScan: A new tool for meta-genomic viral community profiling. BMC Genom. 2016, 17, 165. [Google Scholar] [CrossRef]
- Tithi, S.S.; Aylward, F.; Jensen, R.V.; Zhang, L. FastViromeExplorer: A pipeline for virus and phage identification and abundance profiling in metagenomics data. PeerJ 2018, 6, e4227. [Google Scholar] [CrossRef] [PubMed]
- LaPierre, N.; Mangul, S.; Alser, M.; Mandric, I.; Wu, N.C.; Koslicki, D.; Eskin, E. MiCoP: Microbial community profiling method for detecting viral and fungal organisms in metagenomic samples. BMC Genom. 2019, 20, 423. [Google Scholar] [CrossRef] [PubMed]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Weynberg, K.; Botté, E.; van Oppen, M.; Webster, N.; Rattei, T. HoloVir: A Workflow for Investigating the Diversity and Function of Viruses in Invertebrate Holobionts. Front. Microbiol. 2016, 7, 822. [Google Scholar] [CrossRef]
- Garretto, A.; Hatzopoulos, T.; Putonti, C. virMine: Automated detection of viral sequences from complex metagenomic samples. PeerJ 2019, 7, e6695. [Google Scholar] [CrossRef]
- Lefebvre, M.; Theil, S.; Ma, Y.; Candresse, T. The VirAnnot Pipeline: A Resource for Automated Viral Diversity Estimation and Operational Taxonomy Units Assignation for Virome Sequencing Data. Phytobiomes J. 2019, 3, 256–259. [Google Scholar] [CrossRef]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from meta-genomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Tamisier, L.; Haegeman, A.; Foucart, Y.; Fouillien, N.; Al Rwahnih, M.; Buzkan, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Lefebvre, M.; et al. Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection. Peer Community J. 2021, 1, e53. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Bobay, L.-M.; Ochman, H. Biological species in the viral world. Proc. Natl. Acad. Sci. USA 2018, 115, 6040–6045. [Google Scholar] [CrossRef] [PubMed]
- Pervez, M.T.; Babar, M.E.; Nadeem, A.; Aslam, M.; Awan, A.R.; Aslam, N.; Hussain, T.; Naveed, N.; Qadri, S.; Waheed, U.; et al. Evaluating the Accuracy and Efficiency of Multiple Sequence Alignment Methods. Evol. Bioinform. 2014, 10, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2017, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLOS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Bigot, T.; Temmam, S.; Perot, P.; Eloit, M. RVDB-prot, a reference viral protein database and its HMM profiles. F1000Res 2019, 8, 530. [Google Scholar] [CrossRef] [PubMed]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Páez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2020, 49, D764–D775. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and ap-plications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Baker, D.N.; Salzberg, S.L. KrakenUniq: Confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic se-quences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Hundt, C.; Hildebrandt, A.; Hankeln, T.; Schmidt, B. MetaCache: Context-aware classification of metagenomic reads using minhashing. Bioinformatics 2017, 33, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Piro, V.C.; Dadi, T.H.; Seiler, E.; Reinert, K.; Renard, B.Y. ganon: Precise metagenomics classification against large and up-to-date sets of reference sequences. Bioinformatics 2020, 36 (Suppl. S1). [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Contributors, SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Wick, R. Badread: Simulation of error-prone long reads. J. Open Source Softw. 2019, 4, 1316. [Google Scholar] [CrossRef]
- Gourlé, H.; Karlsson-Lindsjö, O.; Hayer, J.; Bongcam-Rudloff, E. Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics 2018, 35, 521–522. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Vollmers, J.; Wiegand, S.; Kaster, A.-K. Comparing and Evaluating Metagenome Assembly Tools from a Microbiologist’s Perspective—Not Only Size Matters! PLoS ONE 2017, 12, e0169662. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new ge-nome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Metaviral SPAdes: Assembly of viruses from metagenomic data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.D.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Pro-Tocols Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Collins, P.L.; Olmsted, R.A.; Spriggs, M.K.; Johnson, P.R.; Buckler-White, A.J. Gene overlap and site-specific attenuation of transcription of the viral polymerase L gene of human respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 1987, 84, 5134–5138. [Google Scholar] [CrossRef]
- Bertram, G.; Innes, S.; Minella, O.; Richardson, J.P.; Stansfield, I. Endless possibilities: Translation termination and stop co-don recognition. Microbiology 2001, 147, 255–269. [Google Scholar] [CrossRef]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Dreher, T.W.; Miller, W.A. Translational control in positive strand RNA plant viruses. Virology 2005, 344, 185–197. [Google Scholar] [CrossRef]
- Firth, A.E.; Wills, N.M.; Gesteland, R.F.; Atkins, J.F. Stimulation of stop codon readthrough: Frequent presence of an ex-tended 3’ RNA structural element. Nucleic Acids Res. 2011, 39, 6679–6691. [Google Scholar] [CrossRef]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza Viruses and mRNA Splicing: Doing More with Less. mBio 2014, 5, e00070-14. [Google Scholar] [CrossRef]
- Jaafar, Z.A.; Kieft, J.S. Viral RNA structure-based strategies to manipulate translation. Nat. Rev. Genet. 2018, 17, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Plant Health; Bragard, C.; Dehnen-Schmutz, K.; Gonthier, P.; Jacques, M.A.; Jaques Miret, J.A.; Justesen, A.F.; MacLeod, A.; Magnusson, C.S.; Milonas, P.; et al. Pest categorisation of non-EU viruses and viroids of Cydonia Mill., Malus Mill. and Pyrus L. EFSA J. 2019, 17, e05590. [Google Scholar] [CrossRef] [PubMed]
- Canales, C.; Moran, F.; Olmos, A.; Ruiz-Garcia, A.B. First Detection and Molecular Characterization of Apple Stem Grooving Virus, Apple Chlorotic Leaf Spot Virus, and Apple Hammerhead Viroid in Loquat in Spain. Plants 2021, 10, 2293. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Genomes | Unique Contigs | Representative Proteins |
|---|---|---|---|
| Fungus | 2523 | 14,840,506 | 8,485,622 |
| Invertebrate | 1137 | 136,042,698 | 5,593,319 |
| Plant | 769 | 100,686,597 | 6,412,958 |
| Protozoa | 465 | 4,690,439 | 1,701,139 |
| Vertebrate (mammal) | 536 | 198,838,325 | 3,611,964 |
| Vertebrate (other) | 793 | 52,609,300 | 5,459,166 |
| Prokaryote | 31,910 | 3,357,372 | 63,089,651 |
| Sequence Type | Clustering Method | Lineage Groups | Sequences | Database Name |
|---|---|---|---|---|
| Nucleotide (genome) | Unique contigs | 17,231 | 27,029 | genome_nt |
| Nucleotide (nt) | Species, 90% | 40,114 | 181,612 | nt_derep1 |
| Truncated species, 90% | 14,630 | 157,271 | nt_derep2 | |
| Truncated species, 97% | 14,630 | 559,726 | nt_derep3 | |
| Truncated species, 99% | 14,630 | 1,076,325 | nt_derep4 | |
| Protein (genome) | Unique sequences | 17,231 | 686,559 | genome_prot |
| Protein (nr) | Species, 90% | 38,457 | 2,924,049 | nr_derep1 |
| Truncated species, 90% | 14,542 | 2,198,163 | nr_derep2 |
| Method | Database Name | Initial Clusters | Non-Singleton Clusters | Filtered Clusters |
|---|---|---|---|---|
| usearch (50%) | usearch_50 | 253,379 | 90,165 | 6612 |
| usearch (60%) | usearch_60 | 283,612 | 93,286 | 6802 |
| usearch (70%) | usearch_70 | 315,140 | 94,825 | 7310 |
| usearch (80%) | usearch_80 | 350,574 | 94,751 | 6514 |
| usearch (90%) | usearch_90 | 400,303 | 91,950 | 5918 |
| mcl (I = 1.4) * | mcl_1.4 | 135,206 | 58,272 | 397 * |
| mcl (I = 4.0) | mcl_4.0 | 154,114 | 77,032 | 5950 |
| Mixed (usearch 50%) | mixed_50 | 361,297 | 110,525 | 8569 |
| Mixed (usearch 60%) | mixed_60 | 396,879 | 119,293 | 9468 |
| Mixed (usearch 70%) | mixed_70 | 433,833 | 126,782 | 10,283 |
| Mixed (usearch 80%) | mixed_80 | 475,194 | 133,293 | 10,519 |
| Mixed (usearch 90%) | mixed_90 | 532,477 | 139,738 | 9498 |
| Database | Plant Sensitivity | Specificity | F1 Score | Animal Sensitivity | Specificity | F1 Score | Average Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
| usearch_50 | 0.9795 | 0.8895 | 0.9323 | 0.9401 | 0.8869 | 0.9127 | 0.9598 | 0.8882 |
| mcl_1.4 | 0.949 | 0.9285 | 0.9386 | 0.8464 | 0.9022 | 0.8734 | 0.8977 | 0.9153 |
| mcl_4.0 | 0.9822 | 0.8679 | 0.9215 | 0.9905 | 0.8541 | 0.9172 | 0.9864 | 0.861 |
| mixed_90 | 0.9824 | 0.8468 | 0.9096 | 0.9903 | 0.8152 | 0.8943 | 0.9864 | 0.831 |
| Tool | Database | Plant | Vertebrate | Invertebrate | Other |
|---|---|---|---|---|---|
| DeepVirFinder | - | 0.6654 | 0.6039 | 0.7223 | 0.9104 |
| FastViromeExplorer | eukvir | 0.0279 | 0.0102 | 0.0064 | 0.0030 |
| gov | 0.0000 | 0.0000 | 0.0011 | 0.0002 | |
| imgvr | 0.0000 | 0.0000 | 0.0000 | 0.0000 | |
| ncbi | 0.0401 | 0.0138 | 0.0070 | 0.0115 | |
| MiCOP | - | 0.8875 | 0.7458 | 0.8947 | 0.8073 |
| VirFinder | default | 0.4442 | 0.3654 | 0.4629 | 0.8437 |
| modEPV | 0.9149 | 0.8083 | 0.8668 | 0.8539 | |
| ViromeScan | - | 0.9149 | 0.8699 | 0.5994 | 0.7890 |
| VirSorter | db1 | 0.0098 | 0.0978 | 0.0199 | 0.7397 |
| db2 | 0.0098 | 0.0943 | 0.0199 | 0.7316 | |
| VirSorter2 | - | 0.8664 | 0.9411 | 0.9635 | 0.9843 |
| VirTool | - | 0.8855 | 0.0063 | 0.0086 | 0.0093 |
| Host | Library Type | Platform | Assembler | Prodigal | TransDecoder | TransDecoder (Guided) |
|---|---|---|---|---|---|---|
| Retained | DNA | ONT | - | 24 | 14 | 24 |
| HiSeq | MegaHIT | 24 | 24 | 24 | ||
| SPAdes (meta) | 21 | 21 | 17 | |||
| SPAdes (metaviral) | 24 | 24 | 24 | |||
| RNA | ONT | - | 141 | 141 | 140 | |
| HiSeq | MegaHIT | 141 | 141 | 141 | ||
| SPAdes (rna) | 141 | 141 | 141 | |||
| SPAdes (rnaviral) | 141 | 141 | 141 | |||
| Removed | DNA | ONT | - | 24 | 24 | 24 |
| HiSeq | MegaHIT | 24 | 14 | 14 | ||
| SPAdes (meta) | 24 | 17 | 17 | |||
| SPAdes (metaviral) | 0 | - | - | |||
| RNA | ONT | - | 141 | 113 | 113 | |
| HiSeq | MegaHIT | 141 | 130 | 130 | ||
| SPAdes (rna) | 141 | 140 | 140 | |||
| SPAdes (rnaviral) | 141 | 133 | 133 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waite, D.W.; Liefting, L.; Delmiglio, C.; Chernyavtseva, A.; Ha, H.J.; Thompson, J.R. Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity. Viruses 2022, 14, 2163. https://doi.org/10.3390/v14102163
Waite DW, Liefting L, Delmiglio C, Chernyavtseva A, Ha HJ, Thompson JR. Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity. Viruses. 2022; 14(10):2163. https://doi.org/10.3390/v14102163
Chicago/Turabian StyleWaite, David W., Lia Liefting, Catia Delmiglio, Anastasia Chernyavtseva, Hye Jeong Ha, and Jeremy R. Thompson. 2022. "Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity" Viruses 14, no. 10: 2163. https://doi.org/10.3390/v14102163
APA StyleWaite, D. W., Liefting, L., Delmiglio, C., Chernyavtseva, A., Ha, H. J., & Thompson, J. R. (2022). Development and Validation of a Bioinformatic Workflow for the Rapid Detection of Viruses in Biosecurity. Viruses, 14(10), 2163. https://doi.org/10.3390/v14102163