
Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model

 , ,
, ,
Abstract
1. Introduction
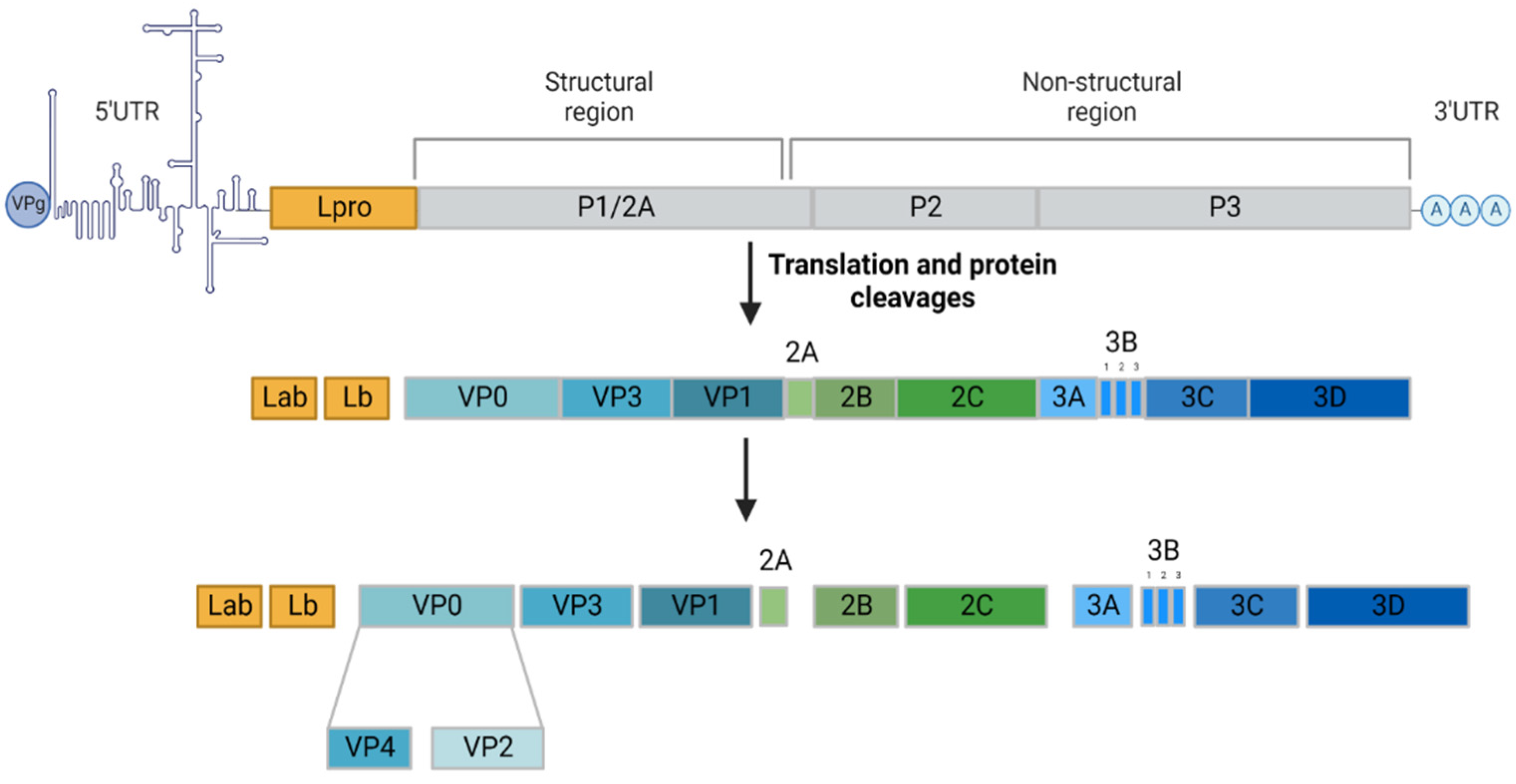
2. FMDV Life Cycle
3. FMDV Persistence in Its Hosts
4. FMDV and Innate Immunity
4.1. Lpro
4.2. 3C
4.3. 3A
4.4. 2B
4.5. VP1
4.6. 2C
4.7. VP3
4.8. VP0
4.9. 3B
4.10. 3D
5. Discussion and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Knowles, N.J.; Samuel, A.R. Molecular Epidemiology of Foot-and-Mouth Disease Virus. Virus Res. 2003, 91, 65–80. [Google Scholar] [CrossRef]
- Belsham, G.J.; Kristensen, T.; Jackson, T. Foot-and-Mouth Disease Virus: Prospects for Using Knowledge of Virus Biology to Improve Control of This Continuing Global Threat. Virus Res. 2020, 281, 197909. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Pacheco, J.M.; Gregg, D.; Baxt, B. Analysis of Foot-and-Mouth Disease Virus Integrin Receptor Expression in Tissues from Naïve and Infected Cattle. J. Comp. Pathol. 2009, 141, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.; Ellard, F.M.; Ghazaleh, R.A.; Brookes, S.M.; Blakemore, W.E.; Corteyn, A.H.; Stuart, D.I.; Newman, J.W.; King, A.M. Efficient Infection of Cells in Culture by Type O Foot-and-Mouth Disease Virus Requires Binding to Cell Surface Heparan Sulfate. J. Virol. 1996, 70, 5282–5287. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A.; González-Magaldi, M.; Sandvig, K.; Sobrino, F.; Armas-Portela, R. Productive Entry of Type C Foot-and-Mouth Disease Virus into Susceptible Cultured Cells Requires Clathrin and Is Dependent on the Presence of Plasma Membrane Cholesterol. Virology 2007, 369, 105–118. [Google Scholar] [CrossRef]
- Vázquez-Calvo, Á.; Saiz, J.-C.; McCullough, K.C.; Sobrino, F.; Martín-Acebes, M.A. Acid-Dependent Viral Entry. Virus Res. 2012, 167, 125–137. [Google Scholar] [CrossRef]
- Groppelli, E.; Levy, H.C.; Sun, E.; Strauss, M.; Nicol, C.; Gold, S.; Zhuang, X.; Tuthill, T.J.; Hogle, J.M.; Rowlands, D.J. Picornavirus RNA Is Protected from Cleavage by Ribonuclease during Virion Uncoating and Transfer across Cellular and Model Membranes. PLoS Pathog. 2017, 13, e1006197. [Google Scholar] [CrossRef]
- Polatnick, J.; Wool, S.H. Association of Foot-and-Mouth Disease Virus Induced RNA Polymerase with Host Cell Organelles. Comp. Immunol. Microbiol. Infect. Dis. 1983, 6, 265–272. [Google Scholar] [CrossRef]
- Loundras, E.-A.; Streetley, J.; Herod, M.R.; Thompson, R.; Harris, M.; Bhella, D.; Stonehouse, N.J. Higher-Order Structures of the Foot-and-Mouth Disease Virus RNA-Dependent RNA Polymerase Required for Genome Replication. Commun. Biol. 2022, 5, 61. [Google Scholar] [CrossRef]
- Grubman, M.J.; Morgan, D.O.; Kendall, J.; Baxt, B. Capsid Intermediates Assembled in a Foot-and-Mouth Disease Virus Genome RNA-Programmed Cell-Free Translation System and in Infected Cells. J. Virol. 1985, 56, 120–126. [Google Scholar] [CrossRef]
- Arzt, J.; Juleff, N.; Zhang, Z.; Rodriguez, L.L. The Pathogenesis of Foot-and-Mouth Disease I: Viral Pathways in Cattle. Transbound. Emerg. Dis. 2011, 58, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Arzt, J.; Baxt, B.; Grubman, M.J.; Jackson, T.; Juleff, N.; Rhyan, J.; Rieder, E.; Waters, R.; Rodriguez, L.L. The Pathogenesis of Foot-and-Mouth Disease II: Viral Pathways in Swine, Small Ruminants, and Wildlife; Myotropism, Chronic Syndromes, and Molecular Virus–Host Interactions. Transbound. Emerg. Dis. 2011, 58, 305–326. [Google Scholar] [CrossRef]
- Sutmoller, P.; McVicar, J.W.; Cottral, G.E. The Epizootiological Importance of Foot-and-Mouth Disease Carriers. Arch. Gesamte Virusforsch. 1968, 23, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Stenfeldt, C.; Eschbaumer, M.; Rekant, S.I.; Pacheco, J.M.; Smoliga, G.R.; Hartwig, E.J.; Rodriguez, L.L.; Arzt, J. The Foot-and-Mouth Disease Carrier State Divergence in Cattle. J. Virol. 2016, 90, 6344–6364. [Google Scholar] [CrossRef] [PubMed]
- Burrows, R. The Persistence of Foot-and Mouth Disease Virus in Sheep. J. Hyg. 1968, 66, 633–640. [Google Scholar] [CrossRef]
- Stenfeldt, C.; Pacheco, J.M.; Smoliga, G.R.; Bishop, E.; Pauszek, S.J.; Hartwig, E.J.; Rodriguez, L.L.; Arzt, J. Detection of Foot-and-Mouth Disease Virus RNA and Capsid Protein in Lymphoid Tissues of Convalescent Pigs Does Not Indicate Existence of a Carrier State. Transbound. Emerg. Dis. 2016, 63, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M.; Smoliga, G.R.; O’Donnell, V.; Brito, B.P.; Stenfeldt, C.; Rodriguez, L.L.; Arzt, J. Persistent Foot-and-Mouth Disease Virus Infection in the Nasopharynx of Cattle; Tissue-Specific Distribution and Local Cytokine Expression. PLoS ONE 2015, 10, e0125698. [Google Scholar] [CrossRef]
- Stenfeldt, C.; Pacheco, J.M.; Singanallur, N.B.; Vosloo, W.; Rodriguez, L.L.; Arzt, J. Virulence beneath the Fleece; a Tale of Foot-and-Mouth Disease Virus Pathogenesis in Sheep. PLoS ONE 2019, 14, e0227061. [Google Scholar] [CrossRef] [PubMed]
- Maree, F.; de Klerk-Lorist, L.-M.; Gubbins, S.; Zhang, F.; Seago, J.; Pérez-Martín, E.; Reid, L.; Scott, K.; van Schalkwyk, L.; Bengis, R.; et al. Differential Persistence of Foot-and-Mouth Disease Virus in African Buffalo Is Related to Virus Virulence. J. Virol. 2016, 90, 5132–5140. [Google Scholar] [CrossRef]
- Arzt, J.; Fish, I.H.; Bertram, M.R.; Smoliga, G.R.; Hartwig, E.J.; Pauszek, S.J.; Holinka-Patterson, L.; Segundo, F.C.D.-S.; Sitt, T.; Rieder, E.; et al. Simultaneous and Staggered Foot-and-Mouth Disease Virus Coinfection of Cattle. J. Virol. 2021, 95, e01650-21. [Google Scholar] [CrossRef]
- Ferretti, L.; Pérez-Martín, E.; Zhang, F.; Maree, F.; de Klerk-Lorist, L.-M.; van Schalkwykc, L.; Juleff, N.D.; Charleston, B.; Ribeca, P. Pervasive Within-Host Recombination and Epistasis as Major Determinants of the Molecular Evolution of the Foot-and-Mouth Disease Virus Capsid. PLoS Pathog. 2020, 16, e1008235. [Google Scholar] [CrossRef]
- De la Torre, J.C.; Martínez-Salas, E.; Diez, J.; Villaverde, A.; Gebauer, F.; Rocha, E.; Dávila, M.; Domingo, E. Coevolution of Cells and Viruses in a Persistent Infection of Foot-and-Mouth Disease Virus in Cell Culture. J. Virol. 1988, 62, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Díez, J.; Dávila, M.; Escarmís, C.; Mateu, M.G.; Dominguez, J.; Pérez, J.J.; Giralt, E.; Melero, J.A.; Domingo, E. Unique Amino Acid Substitutions in the Capsid Proteins of Foot-and-Mouth Disease Virus from a Persistent Infection in Cell Culture. J. Virol. 1990, 64, 5519–5528. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Pacheco, J.M.; Larocco, M.; Gladue, D.P.; Pauszek, S.J.; Smoliga, G.; Krug, P.W.; Baxt, B.; Borca, M.V.; Rodriguez, L. Virus–Host Interactions in Persistently FMDV-Infected Cells Derived from Bovine Pharynx. Virology 2014, 468–470, 185–196. [Google Scholar] [CrossRef]
- Kopliku, L.; Relmy, A.; Romey, A.; Gorna, K.; Zientara, S.; Bakkali-Kassimi, L.; Blaise-Boisseau, S. Establishment of Persistent Foot-and-Mouth Disease Virus (FMDV) Infection in MDBK Cells. Arch. Virol. 2015, 160, 2503–2516. [Google Scholar] [CrossRef]
- Bonjardim, C.A. Interferons (IFNs) Are Key Cytokines in Both Innate and Adaptive Antiviral Immune Responses—And Viruses Counteract IFN Action. Microbes Infect. 2005, 7, 569–578. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- De Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I Interferon Receptors: Biochemistry and Biological Functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of Innate Immune Antiviral Response by NOD2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, E.; Signor, L.; Singh, S.; Boeri Erba, E.; Cusack, S. Structures of the Inactive and Active States of RIP2 Kinase Inform on the Mechanism of Activation. PLoS ONE 2017, 12, e0177161. [Google Scholar] [CrossRef]
- Borrego, B.; Rodríguez-Pulido, M.; Revilla, C.; Álvarez, B.; Sobrino, F.; Domínguez, J.; Sáiz, M. Synthetic RNAs Mimicking Structural Domains in the Foot-and-Mouth Disease Virus Genome Elicit a Broad Innate Immune Response in Porcine Cells Triggered by RIG-I and TLR Activation. Viruses 2015, 7, 3954–3973. [Google Scholar] [CrossRef] [PubMed]
- Lannes, N.; Python, S.; Summerfield, A. Interplay of Foot-and-Mouth Disease Virus, Antibodies and Plasmacytoid Dendritic Cells: Virus Opsonization under Non-Neutralizing Conditions Results in Enhanced Interferon-Alpha Responses. Vet. Res. 2012, 43, 64. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Chen, Y.-P.; Fang, C.-H.; Huang, P.-Y.; Liang, S.-M. Capsid Proteins of Foot-and-Mouth Disease Virus Interact with TLR2 and CD14 to Induce Cytokine Production. Immunol. Lett. 2020, 223, 10–16. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Yang, W.; Wang, Y.; Li, L.; Zheng, H. Foot-and-Mouth Disease Virus VP3 Protein Acts as a Critical Proinflammatory Factor by Promoting Toll-like Receptor 4-Mediated Signaling. J. Virol. 2021, 95, e01120-21. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.; Barber, G. IRF7: Activation, Regulation, Modification and Function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef]
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423. [Google Scholar] [CrossRef]
- Pfeffer, L.M. The Role of Nuclear Factor ΚB in the Interferon Response. J. Interf. Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor Necrosis Factor Receptor Associated Factor 6 (TRAF6) Regulation of Development, Function, and Homeostasis of the Immune System. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Cohen, B.; Rubinstein, M. The Human Interferon Aβ Receptor: Characterization and Molecular Cloning. Cell 1994, 77, 391–400. [Google Scholar] [CrossRef]
- Gauzzi, M.C.; Velazquez, L.; McKendry, R.; Mogensen, K.E.; Fellous, M.; Pellegrini, S. Interferon-α-Dependent Activation of Tyk2 Requires Phosphorylation of Positive Regulatory Tyrosines by Another Kinase. J. Biol. Chem. 1996, 271, 20494–20500. [Google Scholar] [CrossRef] [PubMed]
- Activation of JAK Kinases and STAT Proteins by Interleukin-2 and Interferon Alpha, but Not the T Cell Antigen Receptor, in Human T Lymphocytes. EMBO J. 1994, 13, 5605–5615. [CrossRef]
- Beachboard, D.C.; Horner, S.M. Innate Immune Evasion Strategies of DNA and RNA Viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, S.-Q.; Guo, H.-C. Biological Function of Foot-and-Mouth Disease Virus Non-Structural Proteins and Non-Coding Elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef]
- Medina, G.N.; Segundo, F.D.-S.; Stenfeldt, C.; Arzt, J.; de los Santos, T. The Different Tactics of Foot-and-Mouth Disease Virus to Evade Innate Immunity. Front. Microbiol. 2018, 9, 2644. [Google Scholar] [CrossRef]
- Li, K.; Wang, C.; Yang, F.; Cao, W.; Zhu, Z.; Zheng, H. Virus–Host Interactions in Foot-and-Mouth Disease Virus Infection. Front. Immunol. 2021, 12, 571509. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Chen, T.-C.; Weng, K.-F.; Chang, S.-C.; Chen, L.-L.; Shih, S.-R. Viral and Host Proteins Involved in Picornavirus Life Cycle. J. Biomed. Sci. 2009, 16, 103. [Google Scholar] [CrossRef]
- Jamal, S.M.; Belsham, G.J. Foot-and-Mouth Disease: Past, Present and Future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef]
- Zhu, J.; Weiss, M.; Grubman, M.J. Differential Gene Expression in Bovine Cells Infected with Wild Type and Leaderless Foot-and-Mouth Disease Virus. Virol. J. 2010, 404, 32–40. [Google Scholar] [CrossRef]
- Steinberger, J.; Skern, T. The Leader Proteinase of Foot-and-Mouth Disease Virus: Structure-Function Relationships in a Proteolytic Virulence Factor. Biol. Chem. 2014, 395, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Kirchweger, R.; Ziegler, E.; Lamphear, B.J.; Waters, D.; Liebig, H.D.; Sommergruber, W.; Sobrino, F.; Hohenadl, C.; Blaas, D.; Rhoads, R.E. Foot-and-Mouth Disease Virus Leader Proteinase: Purification of the Lb Form and Determination of Its Cleavage Site on EIF-4 Gamma. J. Virol. 1994, 68, 5677–5684. [Google Scholar] [CrossRef]
- Devaney, M.A.; Vakharia, V.N.; Lloyd, R.E.; Ehrenfeld, E.; Grubman, M.J. Leader Protein of Foot-and-Mouth Disease Virus Is Required for Cleavage of the P220 Component of the Cap-Binding Protein Complex. J. Virol. 1988, 62, 4407–4409. [Google Scholar] [CrossRef] [PubMed]
- Gradi, A.; Foeger, N.; Strong, R.; Svitkin, Y.V.; Sonenberg, N.; Skern, T.; Belsham, G.J. Cleavage of Eukaryotic Translation Initiation Factor 4GII within Foot-and-Mouth Disease Virus-Infected Cells: Identification of the L-Protease Cleavage Site In Vitro. J. Virol. 2004, 78, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles of Its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Martínez-Salas, E.; Ramos, R.; Lafuente, E.; López de Quinto, S. Functional Interactions in Internal Translation Initiation Directed by Viral and Cellular IRES Elements. J. Gen. Virol. 2001, 82, 973–984. [Google Scholar] [CrossRef]
- Rodríguez Pulido, M.; Serrano, P.; Sáiz, M.; Martínez-Salas, E. Foot-and-Mouth Disease Virus Infection Induces Proteolytic Cleavage of PTB, EIF3a,b, and PABP RNA-Binding Proteins. Virology 2007, 364, 466–474. [Google Scholar] [CrossRef]
- Piñeiro, D.; Ramajo, J.; Bradrick, S.S.; Martínez-Salas, E. Gemin5 Proteolysis Reveals a Novel Motif to Identify L Protease Targets. Nucleic Acids Res. 2012, 40, 4942–4953. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. EIF3: A Versatile Scaffold for Translation Initiation Complexes. Trends Biochem. Sci. 2006, 31, 553–562. [Google Scholar] [CrossRef]
- Belsham, G.J.; Brangwyn, J.K. A Region of the 5′ Noncoding Region of Foot-and-Mouth Disease Virus RNA Directs Efficient Internal Initiation of Protein Synthesis within Cells: Involvement with the Role of L Protease in Translational Control. J. Virol. 1990, 64, 5389–5395. [Google Scholar] [CrossRef] [PubMed]
- De los Santos, T.; Diaz-San Segundo, F.; Grubman, M.J. Degradation of Nuclear Factor Kappa B during Foot-and-Mouth Disease Virus Infection. J. Virol. 2007, 81, 12803–12815. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Luo, R.; Ye, R.; Fang, Y.; Xie, L.; Chen, H.; Xiao, S. Foot-and-Mouth Disease Virus Leader Proteinase Inhibits DsRNA-Induced Type I Interferon Transcription by Decreasing Interferon Regulatory Factor 3/7 in Protein Levels. Biochem. Biophys. Res. Commun. 2010, 399, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, M.; Marsili, G.; Remoli, A.L.; Orsatti, R.; Battistini, A. Irf-7 New Role in the Regulation of Genes Involved in Adaptive Immunity. Ann. N. Y. Acad. Sci. 2007, 1095, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, S.; van Pesch, V.; Michiels, T. The Leader Protein of Theiler’s Virus Interferes with Nucleocytoplasmic Trafficking of Cellular Proteins. J. Virol. 2004, 78, 4357–4362. [Google Scholar] [CrossRef]
- Hato, S.V.; Ricour, C.; Schulte, B.M.; Lanke, K.H.W.; de Bruijni, M.; Zoll, J.; Melchers, W.J.G.; Michiels, T.; van Kuppeveld, F.J.M. The Mengovirus Leader Protein Blocks Interferon-α/β Gene Transcription and Inhibits Activation of Interferon Regulatory Factor 3. Cell. Microbiol. 2007, 9, 2921–2930. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Ma, L.; Cai, X.; Xue, Q.; Zheng, H. Seneca Valley Virus 3Cpro Abrogates the IRF3- and IRF7-Mediated Innate Immune Response by Degrading IRF3 and IRF7. Virology 2018, 518, 1–7. [Google Scholar] [CrossRef]
- Medina, G.N.; Knudsen, G.M.; Greninger, A.L.; Kloc, A.; Díaz-San Segundo, F.; Rieder, E.; Grubman, M.J.; DeRisi, J.L.; de los Santos, T. Interaction between FMDV Lpro and Transcription Factor ADNP Is Required for Optimal Viral Replication. Virology 2017, 505, 12–22. [Google Scholar] [CrossRef]
- Quintana, F.J.; Zaltzman, R.; Fernandez-Montesinos, R.; Herrera, J.L.; Gozes, I.; Cohen, I.R.; Pozo, D. NAP, a Peptide Derived from the Activity-Dependent Neuroprotective Protein, Modulates Macrophage Function. Ann. N. Y. Acad. Sci. 2006, 1070, 500–506. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H.; et al. The Leader Proteinase of Foot-and-Mouth Disease Virus Negatively Regulates the Type I Interferon Pathway by Acting as a Viral Deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H. TRIM25 RING-Finger E3 Ubiquitin Ligase Is Essential for RIG-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Pulido, M.R.; Martínez-Salas, E.; Sobrino, F.; Sáiz, M. MDA5 Cleavage by the Leader Protease of Foot-and-Mouth Disease Virus Reveals Its Pleiotropic Effect against the Host Antiviral Response. Cell Death Dis. 2020, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-Catalized Ubiquitination Is Essential for MDA5-Mediated Antiviral Innate Immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.J.; Aloise, C.; Swatek, K.N.; Medina, G.N.; Olek, K.M.; Rabouw, H.H.; de Groot, R.J.; Langereis, M.A.; de los Santos, T.; Komander, D.; et al. Dissecting Distinct Proteolytic Activities of FMDV Lpro Implicates Cleavage and Degradation of RLR Signaling Proteins, Not Its DeISGylase/DUB Activity, in Type I Interferon Suppression. PLoS Pathog. 2020, 16, e1008702. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Pulido, M.; Sánchez-Aparicio, M.T.; Martínez-Salas, E.; García-Sastre, A.; Sobrino, F.; Sáiz, M. Innate Immune Sensor LGP2 Is Cleaved by the Leader Protease of Foot-and-Mouth Disease Virus. PLoS Pathog. 2018, 14, e1007135. [Google Scholar] [CrossRef]
- Murali, A.; Li, X.; Ranjith-Kumar, C.T.; Bhardwaj, K.; Holzenburg, A.; Li, P.; Kao, C.C. Structure and Function of LGP2, a DEX(D/H) Helicase That Regulates the Innate Immunity Response. J. Biol. Chem. 2008, 283, 15825–15833. [Google Scholar] [CrossRef]
- Bruns, A.M.; Horvath, C.M. LGP2 Synergy with MDA5 in RLR-Mediated RNA Recognition and Antiviral Signaling. Cytokine 2015, 74, 198–206. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Liu, L.; Zhong, H.; Chen, Q.; Luo, R.; Liu, X.; Zhang, Z.; Chen, H.; Xiao, S. Foot-and-Mouth Disease Virus (FMDV) Leader Proteinase Negatively Regulates the Porcine Interferon-Λ1 Pathway. Mol. Immunol. 2011, 49, 407–412. [Google Scholar] [CrossRef]
- De los Santos, T.; de Avila Botton, S.; Weiblen, R.; Grubman, M.J. The Leader Proteinase of Foot-and-Mouth Disease Virus Inhibits the Induction of Beta Interferon MRNA and Blocks the Host Innate Immune Response. J. Virol. 2006, 80, 1906–1914. [Google Scholar] [CrossRef]
- Liu, H.; Xue, Q.; Zhu, Z.; Yang, F.; Cao, W.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus Inhibits RIP2 Protein Expression to Promote Viral Replication. Virol. Sin. 2021, 36, 608–622. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in Innate and Adaptive Immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chen, Z.; Lawson, S.R.; Fang, Y. The Cysteine Protease Domain of Porcine Reproductive and Respiratory Syndrome Virus Nonstructural Protein 2 Possesses Deubiquitinating and Interferon Antagonism Functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Xue, Q.; Cai, X.; Liu, X.; Zheng, H. Seneca Valley Virus 3C Protease Negatively Regulates the Type I Interferon Pathway by Acting as a Viral Deubiquitinase. Antivir. Res. 2018, 160, 183–189. [Google Scholar] [CrossRef]
- Andersen, J.; VanScoy, S.; Cheng, T.-F.; Gomez, D.; Reich, N.C. IRF-3-Dependent and Augmented Target Genes during Viral Infection. Genes Immun. 2008, 9, 168–175. [Google Scholar] [CrossRef]
- Li, X.-L.; Blackford, J.A.; Hassel, B.A. RNase L Mediates the Antiviral Effect of Interferon through a Selective Reduction in Viral RNA during Encephalomyocarditis Virus Infection. J. Virol. 1998, 72, 2752–2759. [Google Scholar] [CrossRef]
- Bisbal, C.; Salehzada, T. La RNase L, un acteur essentiel de la réponse cellulaire antivirale. Med. Sci. 2008, 24, 859–864. [Google Scholar] [CrossRef]
- Sui, C.; Jiang, D.; Wu, X.; Liu, S.; Li, F.; Pan, L.; Cong, X.; Li, J.; Yoo, D.; Rock, D.L.; et al. Inhibition of Antiviral Innate Immunity by Foot-and-Mouth Disease Virus Lpro through Interaction with the N-Terminal Domain of Swine RNase L. J. Virol. 2021, 95, e00361-21. [Google Scholar] [CrossRef]
- Kim, S.S.-Y.; Sze, L.; Liu, C.; Lam, K.-P. Correction: The Stress Granule Protein G3BP1 Binds Viral DsRNA and RIG-I to Enhance Interferon-β Response. J. Biol. Chem. 2019, 294, 9655. [Google Scholar] [CrossRef]
- Visser, L.J.; Medina, G.N.; Rabouw, H.H.; de Groot, R.J.; Langereis, M.A.; de los Santos, T.; van Kuppeveld, F.J.M. Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation. J. Virol. 2019, 93, e00922-18. [Google Scholar] [CrossRef] [PubMed]
- Birtley, J.R.; Knox, S.R.; Jaulent, A.M.; Brick, P.; Leatherbarrow, R.J.; Curry, S. Crystal Structure of Foot-and-Mouth Disease Virus 3C Protease: New Insights into Catalytic Mechanism and Cleavage Specificity. J. Biol. Chem. 2005, 280, 11520–11527. [Google Scholar] [CrossRef] [PubMed]
- Palmenberg, A.C. Proteolytic Processing of Picornaviral Polyprotein. Annu. Rev. Microbiol. 1990, 44, 603–623. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, C.; Du, X.; Wang, G.; Cao, W.; Yang, F.; Feng, H.; Zhang, X.; Shi, Z.; Liu, H.; et al. Foot-and-Mouth Disease Virus Infection Inhibits LGP2 Protein Expression to Exaggerate Inflammatory Response and Promote Viral Replication. Cell Death Dis. 2017, 8, e2747. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, A.-Y.; Choi, J.; Park, S.Y.; Park, S.H.; Kim, J.-S.; Lee, S.-I.; Park, J.-H.; Park, C.-K.; Ko, Y.-J. Foot-and-Mouth Disease Virus Evades Innate Immune Response by 3C-Targeting of MDA5. Cells 2021, 10, 271. [Google Scholar] [CrossRef]
- Knox, C.; Moffat, K.; Ali, S.; Ryan, M.; Wileman, T. Foot-and-Mouth Disease Virus Replication Sites Form next to the Nucleus and Close to the Golgi Apparatus, but Exclude Marker Proteins Associated with Host Membrane Compartments. J. Gen. Virol. 2005, 86, 687–696. [Google Scholar] [CrossRef]
- Zhou, Z.; Mogensen, M.M.; Powell, P.P.; Curry, S.; Wileman, T. Foot-and-Mouth Disease Virus 3C Protease Induces Fragmentation of the Golgi Compartment and Blocks Intra-Golgi Transport. J. Virol. 2013, 87, 11721–11729. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Z.; Xue, Q.; Yang, F.; Cao, W.; Zhang, K.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus Antagonizes NOD2-Mediated Antiviral Effects by Inhibiting NOD2 Protein Expression. J. Virol. 2019, 93, e00124-19. [Google Scholar] [CrossRef]
- Abdullah, S.W.; Wu, J.; Zhang, Y.; Bai, M.; Guan, J.; Liu, X.; Sun, S.; Guo, H. DDX21, a Host Restriction Factor of FMDV IRES-Dependent Translation and Replication. Viruses 2021, 13, 1765. [Google Scholar] [CrossRef]
- Calo, E.; Flynn, R.A.; Martin, L.; Spitale, R.C.; Chang, H.Y.; Wysocka, J. RNA Helicase DDX21 Coordinates Transcription and Ribosomal RNA Processing. Nature 2015, 518, 249–253. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.-H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA Helicase Inhibits Influenza A Virus Replication but Is Counteracted by the Viral NS1 Protein. Cell Host Microbe 2014, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Han, T.; Xuan, B.; Sun, Y.; Tang, S.; Yue, N.; Qian, Z. Dissecting the Role of DDX21 in Regulating Human Cytomegalovirus Replication. J. Virol. 2019, 93, e01222-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-Mouth Disease Virus 3C Protease Cleaves NEMO To Impair Innate Immune Signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef]
- Sebban, H.; Yamaoka, S.; Courtois, G. Posttranslational Modifications of NEMO and Its Partners in NF-ΚB Signaling. Trends Cell Biol. 2006, 16, 569–577. [Google Scholar] [CrossRef]
- Yang, F.; Yamashita, J.; Tang, E.; Wang, H.; Guan, K.; Wang, C.-Y. The Zinc Finger Mutation C417R of I-ΚB Kinase γ Impairs Lipopolysaccharide- and TNF-Mediated NF-ΚB Activation through Inhibiting Phosphorylation of the I-ΚB Kinase β Activation Loop. J. Immunol. 2004, 172, 2446–2452. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Wi, S.M.; Shin, D.; Chun, E.; Lee, K.-Y. Peroxiredoxin-6 Negatively Regulates Bactericidal Activity and NF-ΚB Activity by Interrupting TRAF6-ECSIT Complex. Front. Cell. Infect. Microbiol. 2017, 7, 94. [Google Scholar] [CrossRef]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.-K.; Hong, J.T. Roles of Peroxiredoxins in Cancer, Neurodegenerative Diseases and Inflammatory Diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory Functions of Phospholipase A2. Crit. Rev. Immunol. 2017, 37, 127–195. [Google Scholar] [CrossRef]
- Wang, C.; Feng, H.; Zhang, X.; Li, K.; Yang, F.; Cao, W.; Liu, H.; Gao, L.; Xue, Z.; Liu, X.; et al. Porcine Picornavirus 3C Protease Degrades PRDX6 to Impair PRDX6-Mediated Antiviral Function. Virol. Sin. 2021, 36, 948–957. [Google Scholar] [CrossRef]
- He, L.; Zhang, Y.; Lin, Z.; Li, W.; Wang, J.; Li, H.-L. Classical Swine Fever Virus NS5A Protein Localizes to Endoplasmic Reticulum and Induces Oxidative Stress in Vascular Endothelial Cells. Virus Genes 2012, 45, 274–282. [Google Scholar] [CrossRef]
- Belsham, G.J.; McInerney, G.M.; Ross-Smith, N. Foot-and-Mouth Disease Virus 3C Protease Induces Cleavage of Translation Initiation Factors EIF4A and EIF4G within Infected Cells. J. Virol. 2000, 74, 272–280. [Google Scholar] [PubMed]
- Tauber, D.; Tauber, G.; Khong, A.; Van Treeck, B.; Pelletier, J.; Parker, R. Modulation of RNA Condensation by the DEAD-Box Protein EIF4A. Cell 2020, 180, 411–426.e16. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, T.; Rau, M.; Morley, S.J.; Pain, V.M. Proteolytic Cleavage of Initiation Factor EIF-4 Gamma in the Reticulocyte Lysate Inhibits Translation of Capped MRNAs but Enhances That of Uncapped MRNAs. Nucleic Acids Res. 1995, 23, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, P.; Schafer, E.A.; Rieder, E. The Nuclear Protein Sam68 Is Cleaved by the FMDV 3C Protease Redistributing Sam68 to the Cytoplasm during FMDV Infection of Host Cells. Virology 2012, 425, 40–52. [Google Scholar] [CrossRef]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal Protein S3: A KH Domain Subunit in NF-ΚB Complexes That Mediates Selective Gene Regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Liu, Z.-X.; Sun, Y.-P.; Zhu, J.; Lu, S.-Y.; Liu, X.-S.; Huang, Q.-H.; Xie, Y.-Y.; Zhu, H.-B.; Dang, S.-Y.; et al. Rig-I Regulates NF-ΚB Activity through Binding to Nf-Κb1 3′-UTR MRNA. Proc. Natl. Acad. Sci. USA 2013, 110, 6459–6464. [Google Scholar] [CrossRef]
- Guan, J.; Han, S.; Wu, J.; Zhang, Y.; Bai, M.; Abdullah, S.W.; Sun, S.; Guo, H. Ribosomal Protein L13 Participates in Innate Immune Response Induced by Foot-and-Mouth Disease Virus. Front. Immunol. 2021, 12, 616402. [Google Scholar] [CrossRef]
- Falk, M.M.; Grigera, P.R.; Bergmann, I.E.; Zibert, A.; Multhaup, G.; Beck, E. Foot-and-Mouth Disease Virus Protease 3C Induces Specific Proteolytic Cleavage of Host Cell Histone H3. J. Virol. 1990, 64, 748–756. [Google Scholar]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Liu, W.; Yang, D.; Sun, C.; Wang, H.; Zhao, B.; Zhou, G.; Yu, L. HnRNP K Is a Novel Internal Ribosomal Entry Site-Transacting Factor That Negatively Regulates Foot-and-Mouth Disease Virus Translation and Replication and Is Antagonized by Viral 3C Protease. J. Virol. 2020, 94, e00803-20. [Google Scholar] [CrossRef]
- Cao, P.; Luo, W.-W.; Li, C.; Tong, Z.; Zheng, Z.-Q.; Zhou, L.; Xiong, Y.; Li, S. The Heterogeneous Nuclear Ribonucleoprotein HnRNPM Inhibits RNA Virus-Triggered Innate Immunity by Antagonizing RNA Sensing of RIG-I-like Receptors. PLoS Pathog. 2019, 15, e1007983. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E.; Kerr, L.M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R.; et al. 3Cpro of Foot-and-Mouth Disease Virus Antagonizes the Interferon Signaling Pathway by Blocking STAT1/STAT2 Nuclear Translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [PubMed]
- McAllister, C.S.; Taghavi, N.; Samuel, C.E. Protein Kinase PKR Amplification of Interferon β Induction Occurs through Initiation Factor EIF-2α-Mediated Translational Control. J. Biol. Chem. 2012, 287, 36384–36392. [Google Scholar] [CrossRef]
- Bonnet, M.C.; Weil, R.; Dam, E.; Hovanessian, A.G.; Meurs, E.F. PKR Stimulates NF-ΚB Irrespective of Its Kinase Function by Interacting with the IκB Kinase Complex. Mol. Cell. Biol. 2000, 20, 4532–4542. [Google Scholar] [CrossRef]
- Gil, J.; García, M.A.; Gomez-Puertas, P.; Guerra, S.; Rullas, J.; Nakano, H.; Alcamí, J.; Esteban, M. TRAF Family Proteins Link PKR with NF-ΚB Activation. Mol. Cell. Biol. 2004, 24, 4502–4512. [Google Scholar] [CrossRef]
- Li, S.; Min, J.-Y.; Krug, R.M.; Sen, G.C. Binding of the Influenza A Virus NS1 Protein to PKR Mediates the Inhibition of Its Activation by Either PACT or Double-Stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef]
- Vyas, J.; Elia, A.; Clemens, M.J. Inhibition of the Protein Kinase PKR by the Internal Ribosome Entry Site of Hepatitis C Virus Genomic RNA. RNA 2003, 9, 858–870. [Google Scholar] [CrossRef]
- Li, C.; Zhu, Z.; Du, X.; Cao, W.; Yang, F.; Zhang, X.; Feng, H.; Li, D.; Zhang, K.; Liu, X.; et al. Foot-and-Mouth Disease Virus Induces Lysosomal Degradation of Host Protein Kinase PKR by 3C Proteinase to Facilitate Virus Replication. Virology 2017, 509, 222–231. [Google Scholar] [CrossRef]
- Galan, A.; Lozano, G.; Piñeiro, D.; Martinez-Salas, E. G3BP1 Interacts Directly with the FMDV IRES and Negatively Regulates Translation. FEBS J. 2017, 284, 3202–3217. [Google Scholar] [CrossRef]
- Ye, X.; Pan, T.; Wang, D.; Fang, L.; Ma, J.; Zhu, X.; Shi, Y.; Zhang, K.; Zheng, H.; Chen, H.; et al. Foot-and-Mouth Disease Virus Counteracts on Internal Ribosome Entry Site Suppression by G3BP1 and Inhibits G3BP1-Mediated Stress Granule Assembly via Post-Translational Mechanisms. Front. Immunol. 2018, 9, 1142. [Google Scholar] [CrossRef] [PubMed]
- Núñez, J.I.; Baranowski, E.; Molina, N.; Ruiz-Jarabo, C.M.; Sánchez, C.; Domingo, E.; Sobrino, F. A Single Amino Acid Substitution in Nonstructural Protein 3A Can Mediate Adaptation of Foot-and-Mouth Disease Virus to the Guinea Pig. J. Virol. 2001, 75, 3977–3983. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.W.; Mason, P.W. Genetic Determinants of Altered Virulence of Taiwanese Foot-and-Mouth Disease Virus. J. Virol. 2000, 74, 987–991. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Henry, T.M.; O’Donnell, V.K.; Gregory, J.B.; Mason, P.W. Role of Nonstructural Proteins 3A and 3B in Host Range and Pathogenicity of Foot-and-Mouth Disease Virus. J. Virol. 2003, 77, 13017–13027. [Google Scholar] [CrossRef]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative Genomics of Foot-and-Mouth Disease Virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.J.; Davies, P.R.; Henry, T.; O’Donnell, V.; Pacheco, J.M.; Mason, P.W. Emergence in Asia of Foot-and-Mouth Disease Viruses with Altered Host Range: Characterization of Alterations in the 3A Protein. J. Virol. 2001, 75, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Midgley, R.; Moffat, K.; Berryman, S.; Hawes, P.; Simpson, J.; Fullen, D.; Stephens, D.J.; Burman, A.; Jackson, T. A Role for Endoplasmic Reticulum Exit Sites in Foot-and-Mouth Disease Virus Infection. J. Gen. Virol. 2013, 94, 2636–2646. [Google Scholar] [CrossRef]
- Moffat, K.; Howell, G.; Knox, C.; Belsham, G.J.; Monaghan, P.; Ryan, M.D.; Wileman, T. Effects of Foot-and-Mouth Disease Virus Nonstructural Proteins on the Structure and Function of the Early Secretory Pathway: 2BC but Not 3A Blocks Endoplasmic Reticulum-to-Golgi Transport. J. Virol. 2005, 79, 4382–4395. [Google Scholar] [CrossRef]
- Li, D.; Lei, C.; Xu, Z.; Yang, F.; Liu, H.; Zhu, Z.; Li, S.; Liu, X.; Shu, H.; Zheng, H. Foot-and-Mouth Disease Virus Non-Structural Protein 3A Inhibits the Interferon-β Signaling Pathway. Sci. Rep. 2016, 6, 21888. [Google Scholar] [CrossRef]
- Horner, S.M.; Wilkins, C.; Badil, S.; Iskarpatyoti, J.; Gale, M. Proteomic Analysis of Mitochondrial-Associated ER Membranes (MAM) during RNA Virus Infection Reveals Dynamic Changes in Protein and Organelle Trafficking. PLoS ONE 2015, 10, e0117963. [Google Scholar] [CrossRef]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-Associated Endoplasmic Reticulum Membranes (MAM) Form Innate Immune Synapses and Are Targeted by Hepatitis C Virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fu, S.; Wu, Z.; Yang, W.; Ru, Y.; Shu, H.; Liu, X.; Zheng, H. Correction: DDX56 Inhibits Type I Interferon by Disrupting Assembly of IRF3–IPO5 to Inhibit IRF3 Nucleus Import. J. Cell Sci. 2020, 133, jcs244681. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, W.; Ru, Y.; Zhang, K.; Wang, Y.; Liu, X.; Li, D.; Zheng, H. DDX56 Cooperates with FMDV 3A to Enhance FMDV Replication by Inhibiting the Phosphorylation of IRF3. Cell. Signal. 2019, 64, 109393. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, K.; Luo, Z.; Nian, X.; Choudhury, S.K.M.; Zhu, Z.; Song, R.; Pei, J.; Huo, Y.; Li, Y.; et al. FMDV 3A Antagonizes the Effect of ANXA1 to Positively Modulate Viral Replication. J. Virol. 2022, 96, e00317-22. [Google Scholar] [CrossRef]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L.H.K. Annexin-1 Interacts with NEMO and RIP1 to Constitutively Activate IKK Complex and NF-ΚB: Implication in Breast Cancer Metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef]
- Bist, P.; Shu, S.; Lee, H.; Arora, S.; Nair, S.; Lim, J.Y.; Dayalan, J.; Gasser, S.; Biswas, S.K.; Fairhurst, A.-M.; et al. Annexin-A1 Regulates TLR-Mediated IFN-β Production through an Interaction with TANK-Binding Kinase 1. J. Immunol. 2013, 191, 4375–4382. [Google Scholar] [CrossRef]
- Yap, G.L.R.; Sachaphibulkij, K.; Foo, S.L.; Cui, J.; Fairhurst, A.-M.; Lim, L.H.K. Annexin-A1 Promotes RIG-I-Dependent Signaling and Apoptosis via Regulation of the IRF3-IFNAR-STAT1-IFIT1 Pathway in A549 Lung Epithelial Cells. Cell Death Dis. 2020, 11, 463. [Google Scholar] [CrossRef]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R. LRRC25 Inhibits Type I IFN Signaling by Targeting ISG15-associated RIG-I for Autophagic Degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef]
- Yang, W.; Li, D.; Ru, Y.; Bai, J.; Ren, J.; Zhang, J.; Li, L.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus 3A Protein Causes Upregulation of Autophagy-Related Protein LRRC25 To Inhibit the G3BP1-Mediated RIG-like Helicase-Signaling Pathway. J. Virol. 2020, 94, e02086-19. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the Secretory Pathway by Foot-and-Mouth Disease Virus 2BC Protein Is Reproduced by Coexpression of 2B with 2C, and the Site of Inhibition Is Determined by the Subcellular Location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xin, T.; Gao, X.; Wu, J.; Wang, X.; Fang, L.; Sui, X.; Zhu, H.; Cui, S.; Guo, X. Foot-and-Mouth Disease Virus Non-Structural Protein 2B Negatively Regulates the RLR-Mediated IFN-β Induction. Biochem. Biophys. Res. Commun. 2018, 504, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, G.; Yang, F.; Cao, W.; Mao, R.; Du, X.; Zhang, X.; Li, C.; Li, D.; Zhang, K.; et al. Foot-and-Mouth Disease Virus Viroporin 2B Antagonizes RIG-I-Mediated Antiviral Effects by Inhibition of Its Protein Expression. J. Virol. 2016, 90, 11106–11121. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Zheng, W.; Shang, Y.; Zhao, Z.; Wang, S.; Bi, Y.; Zhang, S.; Xu, C.; Duan, Z.; et al. Cyclophilin A-Regulated Ubiquitination Is Critical for RIG-I-Mediated Antiviral Immune Responses. eLife 2017, 6, e24425. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, Q.; Cao, W.; Yang, F.; Ma, L.; Liu, W.; Zhang, K.; Liu, X.; Zhu, Z.; Zheng, H. Foot-and-Mouth Disease Virus Nonstructural Protein 2B Interacts with Cyclophilin A, Modulating Virus Replication. FASEB J. 2018, 32, 6706–6723. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-H.; Zhu, Z.-X.; Cao, W.-J.; Yang, F.; Zhang, X.-L.; Du, X.-L.; Zhang, K.-S.; Liu, X.-T.; Zheng, H.-X. Foot-and-Mouth Disease Virus Induces Lysosomal Degradation of NME1 to Impair P53-Regulated Interferon-Inducible Antiviral Genes Expression. Cell Death Dis. 2018, 9, 885. [Google Scholar] [CrossRef]
- Jung, H.; Seong, H.-A.; Ha, H. Direct Interaction between NM23-H1 and Macrophage Migration Inhibitory Factor (MIF) Is Critical for Alleviation of MIF-Mediated Suppression of P53 Activity. J. Biol. Chem. 2008, 283, 32669–32679. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, H.; Liu, X.; Qi, L.; Gao, X.; Wang, K.; Yao, K.; Zhang, J.; Sun, Y.; Zhang, Y.; et al. Basal Level P53 Suppresses Antiviral Immunity Against Foot-And-Mouth Disease Virus. Viruses 2019, 11, 727. [Google Scholar] [CrossRef]
- Logan, D.; Abu-Ghazaleh, R.; Blakemore, W.; Curry, S.; Jackson, T.; King, A.; Lea, S.; Lewis, R.; Newman, J.; Parry, N.; et al. Structure of a Major Immunogenic Site on Foot-and-Mouth Disease Virus. Nature 1993, 362, 566–568. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Shang, Y.; Zhang, Z.; Liu, X. How Foot-and-Mouth Disease Virus Receptor Mediates Foot-and-Mouth Disease Virus Infection. Virol. J. 2015, 12, 9. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-ΚB Regulation in the Immune System. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, Z.; Lei, Z.; Lei, P. CD14: Biology and Role in the Pathogenesis of Disease. Cytokine Growth Factor Rev. 2019, 48, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory Syncytial Virus Activates Innate Immunity through Toll-Like Receptor 2. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like Receptors 1 and 6 Are Involved in TLR2-Mediated Macrophage Activation by Hepatitis C Virus Core and NS3 Proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef]
- Ekanayaka, P.; Lee, S.-Y.; Herath, T.U.B.; Kim, J.-H.; Kim, T.-H.; Lee, H.; Chathuranga, K.; Chathuranga, W.A.G.; Park, J.-H.; Lee, J.-S. Foot-and-Mouth Disease Virus VP1 Target the MAVS to Inhibit Type-I Interferon Signaling and VP1 E83K Mutation Results in Virus Attenuation. PLoS Pathog. 2020, 16, e1009057. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, W.; Zhang, X.; Wang, C.; Gao, L.; Yang, F.; Cao, W.; Li, K.; Tian, H.; Liu, X.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP1 Interacts with Host Ribosomal Protein SA To Maintain Activation of the MAPK Signal Pathway and Promote Virus Replication. J. Virol. 2020, 94, e01350-19. [Google Scholar] [CrossRef]
- Strong, J.E.; Wong, G.; Jones, S.E.; Grolla, A.; Theriault, S.; Kobinger, G.P.; Feldmann, H. Stimulation of Ebola Virus Production from Persistent Infection through Activation of the Ras/MAPK Pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 17982–17987. [Google Scholar] [CrossRef]
- Mizumura, K.; Hashimoto, S.; Maruoka, S.; Gon, Y.; Kitamura, N.; Matsumoto, K.; Hayashi, S.; Shimizu, K.; Horie, T. Role of Mitogen-Activated Protein Kinases in Influenza Virus Induction of Prostaglandin E2 from Arachidonic Acid in Bronchial Epithelial Cells. Clin. Exp. Allergy 2003, 33, 1244–1251. [Google Scholar] [CrossRef]
- Zhang, K.; Yan, M.; Hao, J.; Shen, C.; Zhu, Z.; Zhang, D.; Hou, J.; Xu, G.; Li, D.; Zheng, H.; et al. Foot-and-Mouth Disease Virus Structural Protein VP1 Destroys the Stability of the TPL2 Trimer by Degradation of TPL2 To Evade Host Antiviral Immunity. J. Virol. 2021, 95, e02149-20. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Liu, J.; Li, Z.; Wang, Y.; Xue, Y.; Li, X.; Cao, H.; Zheng, S.J. Engagement of Soluble Resistance-Related Calcium Binding Protein (Sorcin) with Foot-and-Mouth Disease Virus (FMDV) VP1 Inhibits Type I Interferon Response in Cells. Vet. Microbiol. 2013, 166, 35–46. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Gorbalenya, A.E.; Egger, D.; Bienz, K.; Rinaudo, M.S.; Ehrenfeld, E. Testing the Modularity of the N-Terminal Amphipathic Helix Conserved in Picornavirus 2C Proteins and Hepatitis C NS5A Protein. Virology 2006, 344, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J. Bidirectional Membrane Traffic between the Endoplasmic Reticulum and Golgi Apparatus. Trends Cell Biol. 1993, 3, 81–88. [Google Scholar] [CrossRef]
- Mahajan, S.; Sharma, G.K.; Bora, K.; Pattnaik, B. Identification of Novel Interactions between Host and Non-Structural Protein 2C of Foot-and-Mouth Disease Virus. J. Gen. Virol. 2021, 102, 001577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, X.; Wang, J.; Li, X.; Cao, H.; Wang, Y.; Zeng, Q.; Zheng, S.J. A Critical Role of Interferon-Induced Protein IFP35 in the Type I Interferon Response in Cells Induced by Foot-and-Mouth Disease Virus (FMDV) Protein 2C. Arch. Virol. 2014, 159, 2925–2935. [Google Scholar] [CrossRef]
- Borca, M.V.; Pacheco, J.M.; Holinka, L.G.; Carrillo, C.; Hartwig, E.; Garriga, D.; Kramer, E.; Rodriguez, L.; Piccone, M.E. Role of Arginine-56 within the Structural Protein VP3 of Foot-and-Mouth Disease Virus (FMDV) O1 Campos in Virus Virulence. Virology 2012, 422, 37–45. [Google Scholar] [CrossRef]
- Sa-Carvalho, D.; Rieder, E.; Baxt, B.; Rodarte, R.; Tanuri, A.; Mason, P.W. Tissue Culture Adaptation of Foot-and-Mouth Disease Virus Selects Viruses That Bind to Heparin and Are Attenuated in Cattle. J. Virol. 1997, 71, 5115–5123. [Google Scholar] [CrossRef]
- Rodríguez Pulido, M.; Sobrino, F.; Borrego, B.; Sáiz, M. Attenuated Foot-and-Mouth Disease Virus RNA Carrying a Deletion in the 3′ Noncoding Region Can Elicit Immunity in Swine. J. Virol. 2009, 83, 3475–3485. [Google Scholar] [CrossRef]
- Fry, E.E.; Lea, S.M.; Jackson, T.; Newman, J.W.; Ellard, F.M.; Blakemore, W.E.; Abu-Ghazaleh, R.; Samuel, A.; King, A.M.; Stuart, D.I. The Structure and Function of a Foot-and-Mouth Disease Virus-Oligosaccharide Receptor Complex. EMBO J. 1999, 18, 543. [Google Scholar] [CrossRef]
- Newman, J.; Asfor, A.S.; Berryman, S.; Jackson, T.; Curry, S.; Tuthill, T.J. The Cellular Chaperone Heat Shock Protein 90 Is Required for Foot-and-Mouth Disease Virus Capsid Precursor Processing and Assembly of Capsid Pentamers. J. Virol. 2018, 92, e01415-17. [Google Scholar] [CrossRef]
- Ekanayaka, P.; Lee, B.-H.; Weerawardhana, A.; Chathuranga, K.; Park, J.-H.; Lee, J.-S. Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3. Viruses 2021, 13, 1776. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, W.; Yang, F.; Liu, H.; Zhu, Z.; Lian, K.; Lei, C.; Li, S.; Liu, X.; Zheng, H.; et al. The VP3 Structural Protein of Foot-and-Mouth Disease Virus Inhibits the IFN-β Signaling Pathway. FASEB J. 2016, 30, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wei, J.; Yang, F.; Liu, H.-N.; Zhu, Z.-X.; Cao, W.-J.; Li, S.; Liu, X.-T.; Zheng, H.-X.; Shu, H.-B. Foot-and-Mouth Disease Virus Structural Protein VP3 Degrades Janus Kinase 1 to Inhibit IFN-γ Signal Transduction Pathways. Cell Cycle 2016, 15, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-C.; Guo, H.-C.; Sun, S.-Q. Three-Dimensional Structure of Foot-and-Mouth Disease Virus and Its Biological Functions. Arch. Virol. 2015, 160, 1–16. [Google Scholar] [CrossRef]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 Mediates Degradation of the Adaptor MAVS via the HECT Ubiquitin Ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef]
- Li, D.; Zhang, J.; Yang, W.; He, Y.; Ru, Y.; Fu, S.; Li, L.; Liu, X.; Zheng, H. Poly (RC) Binding Protein 2 Interacts with VP0 and Increases the Replication of the Foot-and-Mouth Disease Virus. Cell Death Dis. 2019, 10, 516. [Google Scholar] [CrossRef]
- Walter, B.L.; Parsley, T.B.; Ehrenfeld, E.; Semler, B.L. Distinct Poly(RC) Binding Protein KH Domain Determinants for Poliovirus Translation Initiation and Viral RNA Replication. J. Virol. 2002, 76, 12008–12022. [Google Scholar] [CrossRef]
- O’Donnell, V.; Pacheco, J.M.; LaRocco, M.; Burrage, T.; Jackson, W.; Rodriguez, L.L.; Borca, M.V.; Baxt, B. Foot-and-Mouth Disease Virus Utilizes an Autophagic Pathway during Viral Replication. Virology 2011, 410, 142–150. [Google Scholar] [CrossRef]
- Nayak, A.; Goodfellow, I.G.; Belsham, G.J. Factors Required for the Uridylylation of the Foot-and-Mouth Disease Virus 3B1, 3B2, and 3B3 Peptides by the RNA-Dependent RNA Polymerase (3Dpol) In Vitro. J. Virol. 2005, 79, 7698–7706. [Google Scholar] [CrossRef]
- Falk, M.M.; Sobrino, F.; Beck, E. VPg Gene Amplification Correlates with Infective Particle Formation in Foot-and-Mouth Disease Virus. J. Virol. 1992, 66, 2251–2260. [Google Scholar] [CrossRef]
- Park, J.-H.; Tark, D.; Lee, K.-N.; Lee, S.-Y.; Ko, M.-K.; Lee, H.-S.; Kim, S.-M.; Ko, Y.-J.; Seo, M.-G.; Chun, J.-E.; et al. Novel Foot-and-Mouth Disease Virus in Korea, July–August 2014. Clin. Exp. Vaccine Res. 2016, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, Z.; Wang, C.; Yang, F.; Cao, W.; Li, P.; Du, X.; Zhao, F.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus 3B Protein Interacts with Pattern Recognition Receptor RIG-I to Block RIG-I–Mediated Immune Signaling and Inhibit Host Antiviral Response. J. Immunol. 2020, 205, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.-W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.H.; Morgan, D.O.; Moore, D.M.; Grubman, M.J.; Card, J.; Fischer, T.; Weddell, G.; Dowbenko, D.; Yansura, D. Identification of Amino Acid and Nucleotide Sequence of the Foot-and-Mouth Disease Virus RNA Polymerase. Virology 1983, 126, 614–623. [Google Scholar] [CrossRef]
- McBride, A.E.; Schlegel, A.; Kirkegaard, K. Human Protein Sam68 Relocalization and Interaction with Poliovirus RNA Polymerase in Infected Cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2296–2301. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.K.; Lawrence, P.; Kloc, A.; Schafer, E.; Rieder, E. Analysis of the Interaction between Host Factor Sam68 and Viral Elements during Foot-and-Mouth Disease Virus Infections. Virol. J. 2015, 12, 224. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, H.; Zeng, Q.; Zheng, H.; Xue, Q.; Cai, X. The DEAD-Box RNA Helicase DDX1 Interacts with the Viral Protein 3D and Inhibits Foot-and-Mouth Disease Virus Replication. Virol. Sin. 2019, 34, 610–617. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; van Kuppeveld, F.J.M. Induction and Suppression of Innate Antiviral Responses by Picornaviruses. Cytokine Growth Factor Rev. 2014, 25, 577–585. [Google Scholar] [CrossRef]
- Kuo, R.-L.; Chen, C.-J.; Wang, R.Y.L.; Huang, H.-I.; Lin, Y.-H.; Tam, E.-H.; Tu, W.-J.; Wu, S.-E.; Shih, S.-R. Role of Enteroviral RNA-Dependent RNA Polymerase in Regulation of MDA5-Mediated Beta Interferon Activation. J. Virol. 2019, 93, e00132-19. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro Targets MDA5 and MAVS in Infected Cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS Mediate Type I Interferon Responses to Coxsackie B Virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, e00546-17. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xie, J.; Xu, S.; Bi, Y.; Li, X.; Zhang, H.; Idris, A.; Bai, J.; Feng, R. Encephalomyocarditis Virus Abrogates the Interferon Beta Signaling Pathway via Its Structural Protein VP2. J. Virol. 2021, 95, e01590-20. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fan, H.; Song, Z.; Liu, X.; Bai, J.; Jiang, P. Encephalomyocarditis Virus 2C Protein Antagonizes Interferon-β Signaling Pathway through Interaction with MDA5. Antivir. Res. 2019, 161, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Drahos, J.; Racaniello, V.R. Cleavage of IPS-1 in Cells Infected with Human Rhinovirus. J. Virol. 2009, 83, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Xue, Y.; Du, J.; Xu, Q.; Yang, X.; Zeng, Y.; Wang, B.; Wang, H.; Liu, J.; Cai, K.; et al. Induction and Suppression of Innate Antiviral Responses by Hepatitis A Virus. Front. Microbiol. 2018, 9, 1865. [Google Scholar] [CrossRef]
- Wen, W.; Yin, M.; Zhang, H.; Liu, T.; Chen, H.; Qian, P.; Hu, J.; Li, X. Seneca Valley Virus 2C and 3C Inhibit Type I Interferon Production by Inducing the Degradation of RIG-I. Virology 2019, 535, 122–129. [Google Scholar] [CrossRef]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca Valley Virus Suppresses Host Type I Interferon Production by Targeting Adaptor Proteins MAVS, TRIF, and TANK for Cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef]
- Hägglund, S.; Laloy, E.; Näslund, K.; Pfaff, F.; Eschbaumer, M.; Romey, A.; Relmy, A.; Rikberg, A.; Svensson, A.; Huet, H.; et al. Model of Persistent Foot-and-mouth Disease Virus Infection in Multilayered Cells Derived from Bovine Dorsal Soft Palate. Transbound. Emerg. Dis. 2020, 67, 133–148. [Google Scholar] [CrossRef]
- Pfaff, F.; Hägglund, S.; Zoli, M.; Blaise-Boisseau, S.; Laloy, E.; Koethe, S.; Zühlke, D.; Riedel, K.; Zientara, S.; Bakkali-Kassimi, L.; et al. Proteogenomics Uncovers Critical Elements of Host Response in Bovine Soft Palate Epithelial Cells Following In Vitro Infection with Foot-And-Mouth Disease Virus. Viruses 2019, 11, 53. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular Machinery for Self-Eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, F.; Zhu, Z.; Yang, Y.; Wang, Z.; Cao, W.; Dang, W.; Li, L.; Mao, R.; Liu, Y.; et al. Cellular DNAJA3, a Novel VP1-Interacting Protein, Inhibits Foot-and-Mouth Disease Virus Replication by Inducing Lysosomal Degradation of VP1 and Attenuating Its Antagonistic Role in the Beta Interferon Signaling Pathway. J. Virol. 2019, 93, e00588-19. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zhang, S.; Qin, X.; Chang, X.; Cui, X.; Li, H.; Zhang, S.; Gao, H.; Wang, P.; Zhang, Z.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP2 Activates the Cellular EIF2S1-ATF4 Pathway and Induces Autophagy via HSPB1. Autophagy 2018, 14, 336–346. [Google Scholar] [CrossRef]
- Gladue, D.P.; O’Donnell, V.; Baker-Branstetter, R.; Holinka, L.G.; Pacheco, J.M.; Fernandez-Sainz, I.; Lu, Z.; Brocchi, E.; Baxt, B.; Piccone, M.E.; et al. Foot-and-Mouth Disease Virus Nonstructural Protein 2C Interacts with Beclin1, Modulating Virus Replication. J. Virol. 2012, 86, 12080–12090. [Google Scholar] [CrossRef]
- Ma, X.; Ling, Y.; Li, P.; Sun, P.; Cao, Y.; Bai, X.; Li, K.; Fu, Y.; Zhang, J.; Li, D.; et al. Cellular Vimentin Interacts with Foot-and-Mouth Disease Virus Nonstructural Protein 3A and Negatively Modulates Viral Replication. J. Virol. 2020, 94, e00273-20. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during Viral Infection—A Double-Edged Sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.-L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef]
- Liu, H.; Xue, Q.; Zeng, Q.; Zhu, Z.; Zheng, H. The Kinase STK3 Interacts with the Viral Structural Protein VP1 and Inhibits Foot-and-Mouth Disease Virus Replication. BioMed Res. Int. 2017, 2017, 2481348. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Protein | Cellular Target | Strategy | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Name | Nature | Viral Strain | Name | Impacted Biological Mechanism | Protein Origin | Action Mechanism | Title | Author |
| Lpro | NS | O1K | LGP2 | Signal transmitting | Human | Cleavage | Innate immune sensor LGP2 is cleaved by the Leader protease of foot-and-mouth disease virus | Rodriguez Pulido (2018) [49] |
| Lpro | NS | A12 | MAVS | Signal transmitting | Swine | N.D | Dissecting distinct proteolytic activities of FMDV Lpro implicates cleavage and degradation of RLR signaling proteins, not its deISGylase/DUB activity, in type I interferon suppression | Visser (2020) [50] |
| Lpro | NS | O1K | MDA5 | Signal transmitting | Human | Cleavage | MDA5 Cleavage by the Leader Protease of Foot-and-Mouth Disease Virus Reveals Its Pleiotropic Effect against the Host Antiviral Response | Pulido (2020) [51] |
| Lpro | NS | O/ES/2001 | RIG-I | Signal transmitting | Human | Cleavage | The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase | Wang (2011) [52] |
| Lpro | NS | O/BY/CHA/2010 | RIP2 | Signal transmitting | Swine | Cleavage | Foot-and-Mouth Disease Virus Inhibits RIP2 Protein Expression to Promote Viral Replication | Liu (2021) [53] |
| Lpro | NS | O/ES/2001 | TRAF3 | Signal transmitting | Human | Deubiquitination | The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase | Wang (2011) [52] |
| Lpro | NS | O/ES/2001 | TRAF6 | Signal transmitting | Human | Deubiquitination | The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase | Wang (2011) [52] |
| Lpro | NS | O/ES/2001 | IRF3 | Transcription | Swine | Cleavage | Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels | Wang (2010) [54] |
| Lpro | NS | O/ES/2001 | IRF7 | Transcription | Swine | Cleavage | Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels | Wang (2010) [54] |
| Lpro | NS | A12 | p65 | Transcription | Goat | Cleavage | Degradation of Nuclear Factor Kappa B during Foot-and-Mouth Disease Virus Infection | De Los Santos (2007) [55] |
| Lpro | NS | A12 | ADNP | Transcription | Human | Regulatory binding | Interaction between FMDV L pro and transcription factor ADNP is required for optimal viral replication | Medina (2017) [56] |
| Lpro | NS | C-S8 | eIF3a | Translation | Human | Cleavage | Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins | Rodriguez Pulido (2007) [57] |
| Lpro | NS | C-S8 | eIF3b | Translation | Human | Cleavage | Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins | Rodriguez Pulido (2007) [57] |
| Lpro | NS | O6 | eIF4GI | Translation | Human | Cleavage | Leader Protein of Foot-and-Mouth Disease Virus Is Required for Cleavage of the p220 Component of the Cap-BindingProteinComplex | Devaney (1988) [58] |
| Lpro | NS | O6 | eIF4GII | Translation | Human | Cleavage | Leader Protein of Foot-and-Mouth Disease Virus Is Required for Cleavage of the p220 Component of the Cap-BindingProteinComplex | Devaney (1988) [58] |
| Lpro | NS | C-S8 | Gemin-5 | Translation | Human | Cleavage | Gemin5 proteolysis reveals a novel motif to identify L protease targets | Pineiro (2012) [59] |
| Lpro | NS | C-S8 | PABP | Translation | Human | Cleavage | Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins | Rodriguez Pulido (2007) [57] |
| Lpro | NS | C-S8 | PTB | Translation | Human | Cleavage | Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins | Rodriguez Pulido (2007) [57] |
| Lpro | NS | A12 | G3BP1 | Response amplification | Human | Cleavage | Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation | Visser (2019) [60] |
| Lpro | NS | A12 | G3BP2 | Response amplification | Human | Cleavage | Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation | Visser (2019) [60] |
| Lpro | NS | O/Tibet/CHA/99 | RNase L | Response amplification | Swine | Regulatory binding | Inhibition of Antiviral Innate Immunity by Foot-and-Mouth Disease Virus Lpro through Interaction with N-terminal Domain of Swine RNase L | Sui (2021) [61] |
| Lpro | NS | O/ES/2001 | TBK1 | N.D | Human | Deubiquitination and Cleavage | The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase | Wang (2011) [52] |
| 3C | NS | O/BY/CHA/2010 | LGP2 | Viral sensing | Swine | Inhibition of protein expression | Foot-and-mouth disease virus infection inhibits LGP2 protein expression to exaggerate inflammatory response and promote viral replication | Zhu (2017) [62] |
| 3C | NS | O/SKR/2000 | MDA5 | Viral sensing | Swine | Inhibition of protein expression | Foot-and-Mouth Disease Virus Evades Innate Immune Response by 3C-Targeting of MDA5 | Kim (2021) [63] |
| 3C | NS | O/BY/CHA/2010 | NOD2 | Viral sensing | Swine | Cleavage | Foot-and-Mouth Disease Virus Antagonizes NOD2-Mediated Antiviral Effects by Inhibiting NOD2 Protein Expression | Liu (2019) [64] |
| 3C | NS | O/BY/CHA/2010 | DDX21 | Viral sensing and Signal transmitting | Swine | Lysosomal degradation | DDX21, a Host Restriction Factor of FMDV IRES-Dependent Translation and Replication | Abdullah (2021) [65] |
| 3C | NS | O/ES/2001 | NEMO | Signal transmitting | Swine | Cleavage | Foot-and-Mouth Disease Virus 3C Protease Cleaves NEMO To Impair Innate Immune Signaling | Wang (2012) [66] |
| 3C | NS | O/BY/CHA/2010 | RIP2 | Signal transmitting | Swine | Cleavage | Foot-and-Mouth Disease Virus Inhibits RIP2 Protein Expression to Promote Viral Replication | Liu (2021) [53] |
| 3C | NS | O/BY/CHA/2010 | PRDX6 | Signal transmitting | Swine | Cleavage | Porcine Picornavirus 3C Protease Degrades PRDX6 to Impair PRDX6-mediated Antiviral Function | Wang (2021) [67] |
| 3C | NS | O1K | H3 | Transcription | Cattle | Cleavage | Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3 | Falk (1990) [68] |
| 3C | NS | CS8 | eIF4A | Translation | Hamster | Cleavage | Foot-and-Mouth Disease Virus 3C Protease Induces Cleavage of Translation Initiation Factors eIF4A and eIF4G within Infected Cells | Belsham (2000) [69] |
| 3C | NS | CS8 | eIF4G | Translation | Hamster | Cleavage | Foot-and-Mouth Disease Virus 3C Protease Induces Cleavage of Translation Initiation Factors eIF4A and eIF4G within Infected Cells | Belsham (2000) [69] |
| 3C | NS | O/YS/CHA/05 | hnRNP K | Translation | Hamster | Cleavage | hnRNP K Is a Novel Internal Ribosomal Entry Site-Transacting Factor That Negatively Regulates Foot-and-Mouth Disease Virus Translation and Replication and Is Antagonized by Viral 3C Protease | Liu (2020) [70] |
| 3C | NS | O/BY/CHA/2010 | RPL13 | Translation | Swine | Cleavage | Ribosomal Protein L13 Participates in Innate Immune Response Induced by Foot-and-Mouth Disease Virus | Guan (2021) [71] |
| 3C | NS | A24/Cruzeiro/Brazil 1955 | Sam68 | Translation | Human | Cleavage | The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells | Lawrence (2012) [72] |
| 3C | NS | O/ES/2001 | G3BP1 | Response amplification | Swine | Cleavage | Foot-and-Mouth Disease Virus Counteracts on Internal Ribosome Entry Site Suppression by G3BP1 and Inhibits G3BP1-Mediated Stress Granule Assembly via Post-Translational Mechanisms | Ye (2018) [73] |
| 3C | vS | O/BY/CHA/2010 | PKR | Response amplification | Swine | Lysosomal degradation | Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication | Li (2017) [74] |
| 3C | NS | O/Tibet/CHA/99 | STAT1 | Response amplification | Human | Prevention of nuclear translocation | 3Cpro of Foot-and-Mouth Disease Virus Antagonizes the Interferon Signaling Pathway by Blocking STAT1/STAT2 Nuclear Translocation | Du (2014) [75] |
| 3C | NS | O/Tibet/CHA/99 | STAT2 | Response amplification | Human | Prevention of nuclear translocation | 3Cpro of Foot-and-Mouth Disease Virus Antagonizes the Interferon Signaling Pathway by Blocking STAT1/STAT2 Nuclear Translocation | Du (2014) [75] |
| 3A | NS | O/Tibet/CHA/99 | RIG-I | Viral sensing | Human | Regulatory binding and reduction of RNA levels | Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway | Li (2016) [76] |
| 3A | NS | type-O | LRRC25 | Signal transmitting | Swine | Protein upregulation | Foot-and-Mouth Disease Virus 3A Protein Causes Upregulation of Autophagy-Related Protein LRRC25 To Inhibit the G3BP1-Mediated RIG-like Helicase-Signaling Pathway | Yang (2020) [77] |
| 3A | NS | O/Tibet/CHA/99 | MAVS | Signal transmitting | Human | Regulatory binding and reduction of RNA levels | Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway | Li (2016) [76] |
| 3A | NS | O/Tibet/CHA/99 | MDA5 | Signal transmitting | Human | Regulatory binding and reduction of RNA levels | Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway | Li (2016) [76] |
| 3A | NS | O/BY/CHA/2010 | ANXA1 | Signal transmitting | Swine | Regulatory binding | FMDV 3A Antagonizes the Effect of ANXA1 to Positively Modulate Viral Replication | Ma (2022) [78] |
| 3A | NS | O/BY/CHA/2010 | DDX56 | Translation | Swine | Regulatory binding | DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3 | Fu (2019) [79] |
| 2B | NS | O/BY/CHA/2010 | DDX21 | Viral sensing | Swine | N.D | DDX21, a Host Restriction Factor of FMDV IRES-Dependent Translation and Replication | Abdullah (2021) [65] |
| 2B | NS | O/Tibet/CHA/2010 | LGP2 | Viral sensing | Swine | N.D | Foot-and-mouth disease virus infection inhibits LGP2 protein expression to exaggerate inflammatory response and promote viral replication | Zhu (2017) [62] |
| 2B | NS | O/MYA/01/1998 | MDA5 | Viral sensing | Human | Regulatory binding | Foot-and-mouth disease virus non-structural protein 2B negatively regulates the RLR-mediated IFN-β induction | Li (2018) [80] |
| 2B | NS | O/BY/CHA/2010 | NOD2 | Viral sensing | Swine | N.D | Foot-and-Mouth Disease Virus Antagonizes NOD2-Mediated Antiviral Effects by Inhibiting NOD2 Protein Expression | Liu (2019) [64] |
| 2B | NS | O/BY/CHA/2010 | RIG-I | Viral sensing | Swine | Inhibition of protein expression | Foot-and-Mouth Disease Virus Viroporin 2B Antagonizes RIG-I-Mediated Antiviral Effects by Inhibition of Its Protein Expression | Zhu (2016) [81] |
| 2B | NS | O/MYA/01/1998 | RIG-I | Viral sensing | Human | Regulatory binding | Foot-and-mouth disease virus non-structural protein 2B negatively regulates the RLR-mediated IFN-β induction | Li (2018) [80] |
| 2B | NS | O/BY/CHA/2010 | Cyclophilin A | Signal transmitting | Swine | N.D | Foot-and-mouth disease virus nonstructural protein 2B interacts with cyclophilin A, modulating virus replication | Liu (2018) [82] |
| 2B | NS | O/BY/CHA/2010 | RIP2 | Signal transmitting | Swine | N.D | Foot-and-Mouth Disease Virus Inhibits RIP2 Protein Expression to Promote Viral Replication | Liu (2021) [53] |
| 2B | NS | O/BY/CHA/2010 | NME1 | Transcription | Swine | N.D | Foot-and-mouth disease virus induces lysosomal degradation of NME1 to impair p53-regulated interferon-inducible antiviral genes expression | Feng (2018) [83] |
| VP1 | VP | O/Taiwan/97 | TLR2 | Viral sensing | Human | Regulatory binding | Capsid proteins of foot-and-mouth disease virus interact with TLR2 and T CD14 to induce cytokine production | Lin (2020) [36] |
| VP1 | VP | O/Taiwan/97 | CD14 | Signal transmitting | Human | Regulatory binding | Capsid proteins of foot-and-mouth disease virus interact with TLR2 and T CD14 to induce cytokine production | Lin (2020) [36] |
| VP1 | VP | O1/Manisa/Turkey/69 | MAVS | Signal transmitting | N.D | Regulatory binding | Foot-and-mouth disease virus VP1 target the MAVS to inhibit type-I interferon signaling and VP1 E83K mutation results in virus attenuation | Ekanayaka (2020) [84] |
| VP1 | VP | O/BY/CHA/2010 | RPSA | Signal transmitting | Swine | Regulatory binding | Foot-and-Mouth Disease Virus Capsid Protein VP1 Interacts with Host Ribosomal Protein SA To Maintain Activation of the MAPK Signal Pathway and Promote Virus Replication | Zhu (2020) [85] |
| VP1 | VP | N.D | Sorcin | Signal transmitting | Swine | N.D | Engagement of soluble resistance-related calcium binding protein (sorcin) with foot-and-mouth disease virus (FMDV) VP1 inhibits type I interferon response in cells | Li (2013) [86] |
| VP1 | VP | O/BY/CHA/2010 | TPL2 | Signal transmitting | Swine | Polyubiquitination | Foot-and-Mouth Disease Virus Structural Protein VP1 Destroys the Stability of TPL2 Trimer by Degradation TPL2 to Evade Host Antiviral Immunity | Zhang (2020) [87] |
| 2C | NS | O/BY/CHA/2010 | NOD2 | Viral sensing | Swine | Inhibition of protein expression | Foot-and-Mouth Disease Virus Antagonizes NOD2-Mediated Antiviral Effects by Inhibiting NOD2 Protein Expression | Liu (2019) [64] |
| 2C | NS | O/BY/CHA/2010 | DDX21 | Viral sensing and Signal transmitting | Swine | Degradation via caspase pathway | DDX21, a Host Restriction Factor of FMDV IRES-Dependent Translation and Replication | Abdullah (2021) [65] |
| 2C | NS | A/IND40/2000 | MARCHF7 | Signal transmitting | Swine | N.D | Identification of novel interactions between host and non-structural protein 2C of foot-and-mouth disease virus | Mahajan (2021) [88] |
| 2C | NS | N.D | Nmi | Signal transmitting | Swine | Recruitment | A critical role of interferon-induced protein IFP35 in the type I interferon response in cells induced by foot-and-mouth disease virus (FMDV) protein 2C | Zeng (2014) [89] |
| 2C | NS | O/BY/CHA/2010 | RIP2 | Signal transmitting | Swine | Inhibition of protein expression | Foot-and-Mouth Disease Virus Inhibits RIP2 Protein Expression to Promote Viral Replication | Liu (2021) [53] |
| 2C | NS | N.D | IFP35 | Signal transmitting and Transcription | Swine | Recruitment | A critical role of interferon-induced protein IFP35 in the type I interferon response in cells induced by foot-and-mouth disease virus (FMDV) protein 2C | Zeng (2014) [89] |
| VP3 | VP | O/Taiwan/97 | TLR2 | Viral sensing | Human | Regulatory binding | Capsid proteins of foot-and-mouth disease virus interact with TLR2 and T CD14 to induce cytokine production | Lin (2020) [36] |
| VP3 | VP | O/Taiwan/97 | CD14 | Signal transmitting | Human | Regulatory binding | Capsid proteins of foot-and-mouth disease virus interact with TLR2 and T CD14 to induce cytokine production | Lin (2020) [36] |
| VP3 | VP | O/Tibet/CHA/2010 | MAVS | Signal transmitting | Human | Inhibition of mRNA synthesis | The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-β signaling pathway | Li (2016) [90] |
| VP3 | VP | O/Tibet/CHA/99 | JAK 1 | Response amplification | Human | Regulatory binding | Foot-and-mouth disease virus structural protein VP3 degrades Janus kinase 1 to inhibit IFN-γ signal transduction pathways | Li (2016) [91] |
| VP3 | VP | O/Tibet/CHA/99 | JAK 2 | Response amplification | Human | Regulatory binding | Foot-and-mouth disease virus structural protein VP3 degrades Janus kinase 1 to inhibit IFN-γ signal transduction pathways | Li (2016) [91] |
| VP0 | VP | O/Tibet/CHA/99 | PCBP2 | Signal transmitting | Swine | Regulatory binding | Poly (rC) binding protein 2 interacts with VP0 and increases the replication of the foot-and-mouth disease virus | Li (2019) [92] |
| VP4 | VP | O/BY/CHA/2010 | NME1 | Signal transmitting | Swine | Degradation via macroautophagy pathway | Foot-and-mouth disease virus induces lysosomal degradation of NME1 to impair p53-regulated interferon-inducible antiviral genes expression | Feng (2018) [83] |
| 3B | NS | O/BY/CHA/2010 | RIG-I | Signal transmitting | Swine | Regulatory binding | Foot-and-Mouth Disease Virus 3B Protein Interacts with Pattern Recognition Receptor RIG-I to Block RIG-I-Mediated Immune Signaling and Inhibit Host Antiviral Response | Zhang (2020) [93] |
| 3D | NS | A24/Cruzeiro/Brazil 1955 | Sam68 | Translation | Mouse | Regulatory binding | Analysis of the interaction between host factor Sam68 and viral elements during foot-and-mouth disease virus infections | Ray (2015) [94] |
| 3D | NS | O/BY/CHA/2010 | DDX1 | N.D | Swine | Regulatory binding | The DEAD-Box RNA Helicase DDX1 Interacts with the Viral Protein 3D and Inhibits Foot-and-Mouth Disease Virus Replication | Xue (2019) [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarry, M.; Vitour, D.; Zientara, S.; Bakkali Kassimi, L.; Blaise-Boisseau, S. Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model. Viruses 2022, 14, 2129. https://doi.org/10.3390/v14102129
Sarry M, Vitour D, Zientara S, Bakkali Kassimi L, Blaise-Boisseau S. Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model. Viruses. 2022; 14(10):2129. https://doi.org/10.3390/v14102129
Chicago/Turabian StyleSarry, Morgan, Damien Vitour, Stephan Zientara, Labib Bakkali Kassimi, and Sandra Blaise-Boisseau. 2022. "Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model" Viruses 14, no. 10: 2129. https://doi.org/10.3390/v14102129
APA StyleSarry, M., Vitour, D., Zientara, S., Bakkali Kassimi, L., & Blaise-Boisseau, S. (2022). Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model. Viruses, 14(10), 2129. https://doi.org/10.3390/v14102129