
Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
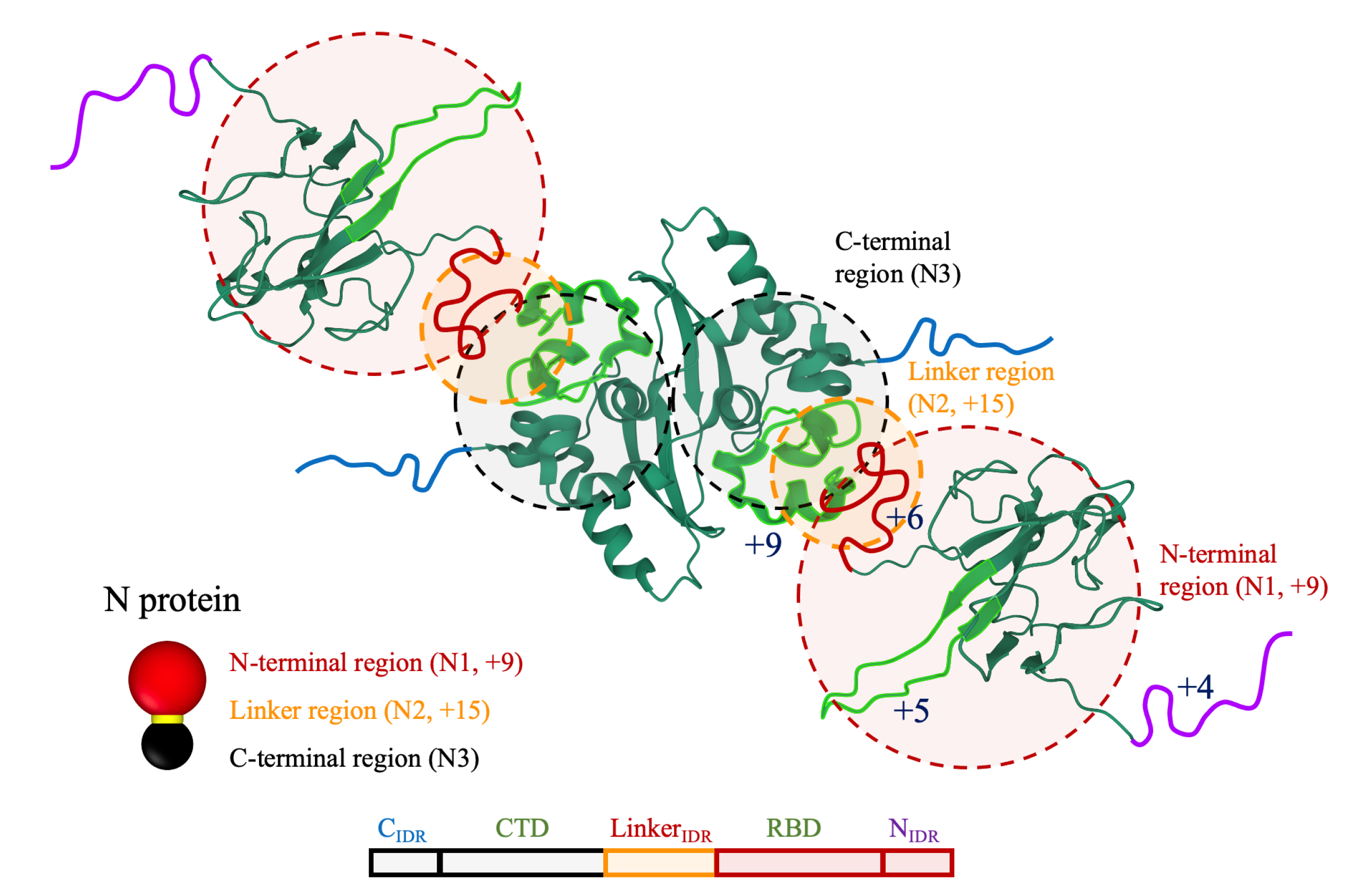
2.1. N Protein
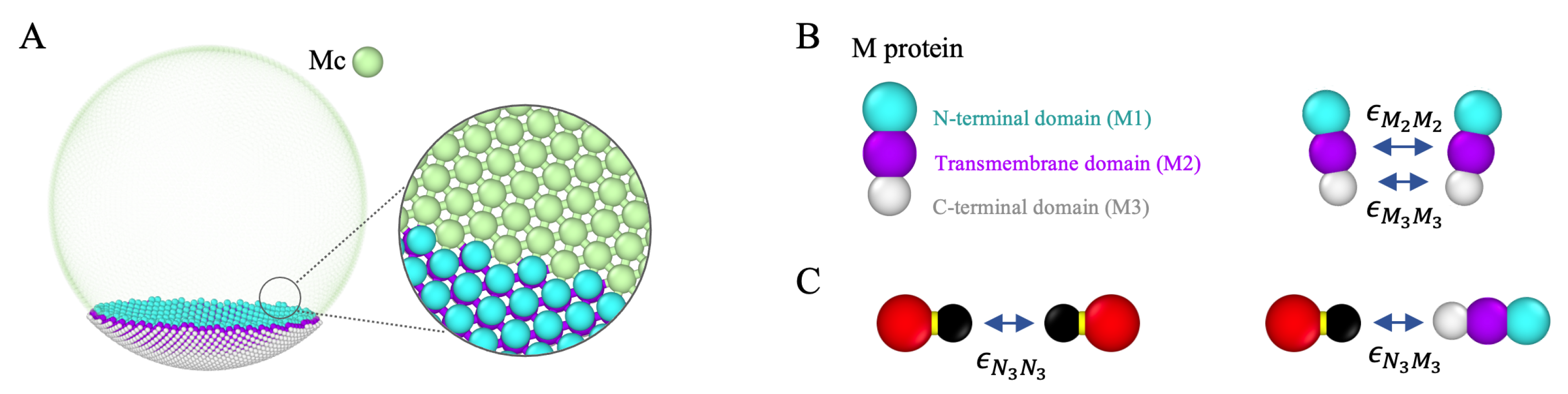
2.2. Membrane and Membrane Proteins
2.3. Genome
2.4. Simulations
3. Results
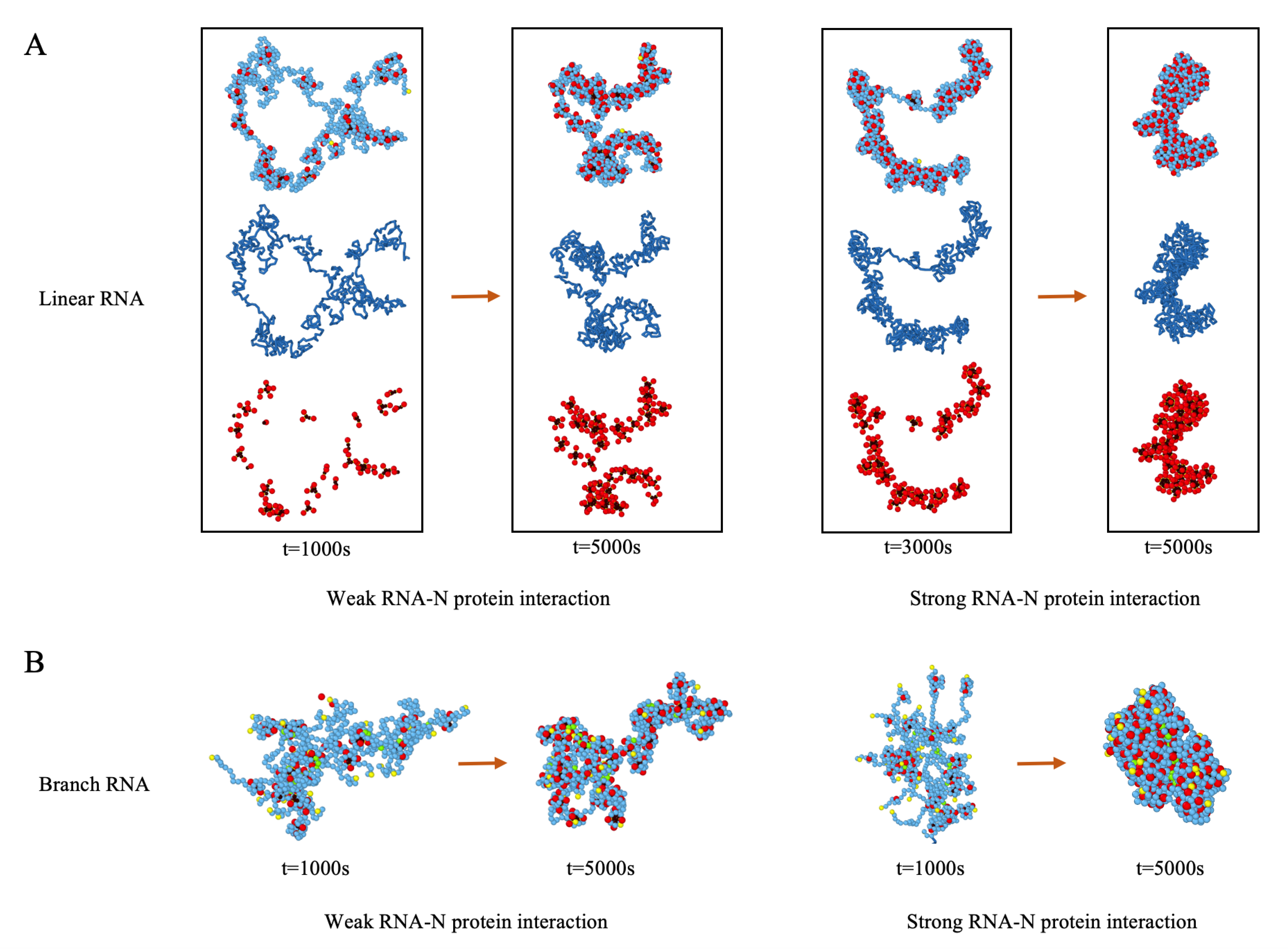
3.1. N Protein and RNA Assemble into RNP with Multiple Protein Clusters
3.2. Spontaneous Radius and Line Tension of M Proteins Are Important for Budding
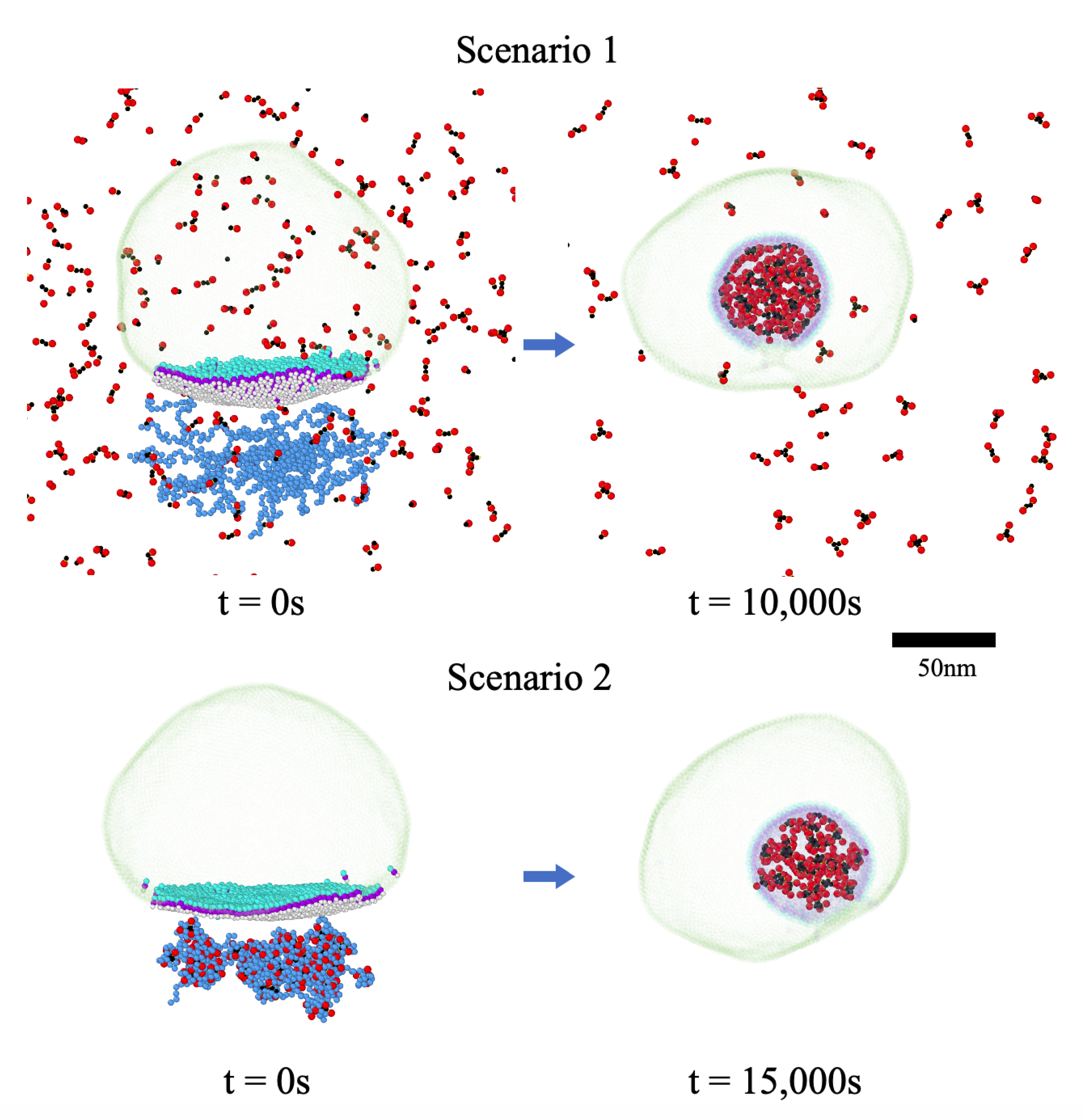
3.3. Two Different Models for RNA Packaging
3.4. Condensation of RNA by N Proteins Is Essential for Its Packaging
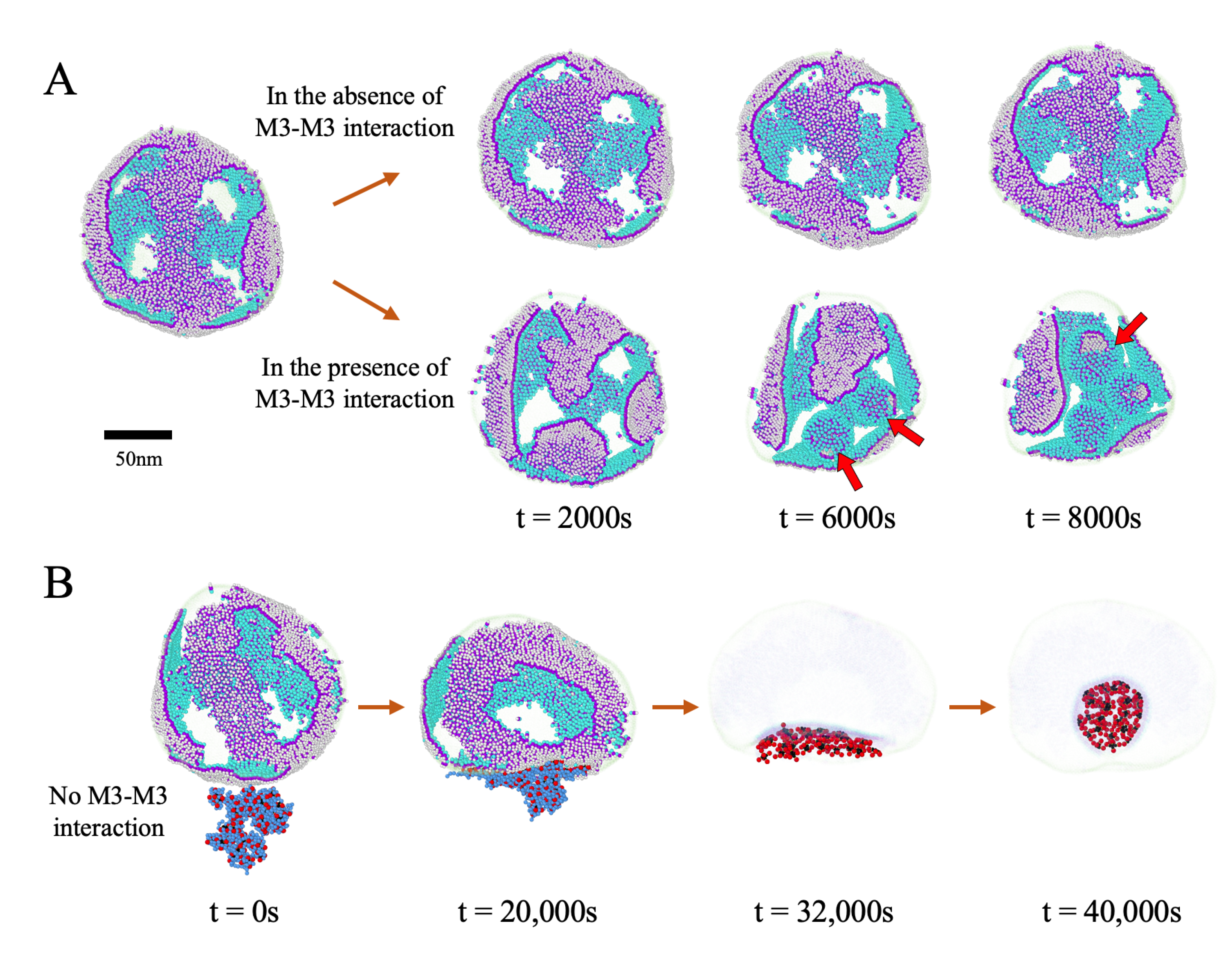
3.5. Impact of the Initial Distribution of M Proteins on Virus Budding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| E protein | Envelope protein |
| M protein | Membrane protein |
| N protein | Nucleocapsid protein |
| S protein | Spike protein |
| RNP | Ribonucleoprotein |
| NPC | N protein clusters |
| ERGIC | Endoplasmic Reticulum-Golgi intermediate compartment |
| VLP | Virus-like particle |
| CG | Coarse-grained |
References
- Chang, C.K.; Hsu, Y.L.; Chang, Y.H.; Chao, F.A.; Wu, M.C.; Huang, Y.S.; Hu, C.K.; Huang, T.H. Multiple Nucleic Acid Binding Sites and Intrinsic Disorder of Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein: Implications for Ribonucleocapsid Protein Packaging. J. Virol. 2009, 83, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Chen, C.M.; Chiang, M.H.; Hsu, Y.L.; Huang, T.H. Transient Oligomerization of the SARS-CoV N Protein—Implication for Virus Ribonucleoprotein Packaging. PLoS ONE 2013, 8, e65045. [Google Scholar] [CrossRef]
- Chang, C.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T. The SARS coronavirus nucleocapsid protein – Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, M.; Veronica Falconieri Hays, B.G.; Christiansen, J. A Visual Guide to the SARS-CoV-2 Coronavirus. Sci. Am. 2020, 323, 32. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.A.; Geuze, H.J.; Slot, J.W.; Klumperman, J. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell 1999, 98, 81–90. [Google Scholar] [CrossRef]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994, 68, 6523–6534. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, M.R.; Davies, H.A.; Nermut, M.V. Ribonucleoprotein-like structures from coronavirus particles. J. Gen. Virol. 1978, 39, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.; Verkleij, A.; Rottier, P.J.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA 2009, 106, 582–587. [Google Scholar] [CrossRef]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Kuo, L.; Masters, P.S. Genetic Evidence for a Structural Interaction between the Carboxy Termini of the Membrane and Nucleocapsid Proteins of Mouse Hepatitis Virus. J. Virol. 2002, 76, 4987–4999. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. Analyses of Coronavirus Assembly Interactions with Interspecies Membrane and Nucleocapsid Protein Chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef]
- Hurst, K.R.; Kuo, L.; Koetzner, C.A.; Ye, R.; Hsue, B.; Masters, P.S. A Major Determinant for Membrane Protein Interaction Localizes to the Carboxy-Terminal Domain of the Mouse Coronavirus Nucleocapsid Protein. J. Virol. 2005, 79, 13285–13297. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, Z.y.; Kong, W.p.; Nabel, G.J. Generation of Synthetic Severe Acute Respiratory Syndrome Coronavirus Pseudoparticles: Implications for Assembly and Vaccine Production. J. Virol. 2004, 78, 12557–12565. [Google Scholar] [CrossRef]
- Arndt, A.L.; Larson, B.J.; Hogue, B.G. A Conserved Domain in the Coronavirus Membrane Protein Tail Is Important for Virus Assembly. J. Virol. 2010, 84, 11418–11428. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.; Buchmeier, M. Supramolecular Architecture of the Coronavirus Particle. Adv. Virus Res. 2020, 105, 93–116. [Google Scholar] [CrossRef]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N Structural Proteins of the Severe Acute Respiratory Syndrome Coronavirus Are Required for Efficient Assembly, Trafficking, and Release of Virus-Like Particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef]
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 viral budding and entry can be modeled using BSL-2 level virus-like particles. J. Biol. Chem. 2021, 296, 100103. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Roy, P.; Travesset, A.; Zandi, R. Why large icosahedral viruses need scaffolding proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10971–10976. [Google Scholar] [CrossRef] [PubMed]
- Panahandeh, S.; Li, S.; Dragnea, B.; Zandi, R. Virus Assembly Pathways Inside a Host Cell. ACS Nano 2022, 16, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Qiao, C.; Hagan, M.F. Viral genome structures are optimal for capsid assembly. eLife 2013, 2013, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, Z.; Glotzer, S.C. A Precise Packing Sequence for Self-Assembled Convex Structures. Proc. Natl. Acad. Sci. USA 2007, 104, 717. [Google Scholar] [CrossRef]
- Fejer, S.; Chakrabarti, D.; Wales, D. Emergent Complexity from Simple Anisotropic Building Blocks: Shells, Tubes, and Spirals. Nano Lett. 2010, 4, 219. [Google Scholar] [CrossRef]
- Rapaport, D.C. Self-assembly of polyhedral shells: A molecular dynamics study. Phys. Rev. E 2004, 70, 051905. [Google Scholar] [CrossRef]
- Patel, N.; Dykeman, E.C.; Coutts, R.H.A.; Lomonossoff, G.P.; Rowlands, D.J.; Phillips, S.E.V.; Ranson, N.; Twarock, R.; Tuma, R.; Stockley, P.G. Revealing the density of encoded functions in a viral RNA. Proc. Natl. Acad. Sci. USA 2015, 112, 2227–2232. [Google Scholar] [CrossRef]
- Vernizzi, G.; Olvera de la Cruz, M. Faceting ionic shells into icosahedra via electrostatics. Proc. Natl. Acad. Sci. USA 2007, 104, 18382–18386. [Google Scholar] [CrossRef]
- Paquay, S.; Kusumaatmaja, H.; Wales, D.J.; Zandi, R.; Van Der Schoot, P. Energetically favoured defects in dense packings of particles on spherical surfaces. Soft Matter 2016, 12, 5708–5717. [Google Scholar] [CrossRef]
- Collins, L.T.; Elkholy, T.; Mubin, S.; Hill, D.; Williams, R.; Ezike, K.; Singhal, A. Elucidation of SARS-Cov-2 Budding Mechanisms through Molecular Dynamics Simulations of M and E Protein Complexes. J. Phys. Chem. Lett. 2021, 12, 12249–12255. [Google Scholar] [CrossRef]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the formation, structure and free energy relationships of M protein dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Hadden, J.A.; Zlotnick, A.; Ou, J.H.J. Assembly Properties of Hepatitis B Virus Core Protein Mutants Correlate with Their Resistance to Assembly-Directed Antivirals. J. Virol. 2018, 92, e01082-18. [Google Scholar] [CrossRef]
- Nair, S.; Li, L.; Francis, S.; Turner, W.W.; VanNieuwenhze, M.; Zlotnick, A. Use of a Fluorescent Analogue of a HBV Core Protein-Directed Drug To Interrogate an Antiviral Mechanism. J. Am. Chem. Soc. 2018, 140, 15261–15269. [Google Scholar] [CrossRef]
- Perlmutter, J.D.; Perkett, M.R.; Hagan, M.F. Pathways for Virus Assembly Around Nucleic Acids. J. Mol. Biol. 2014, 426, 3148–3165. [Google Scholar] [CrossRef] [PubMed]
- Hagan, M.F. Modeling Viral Capsid Assembly. Adv. Chem. Phys. 2014, 155, 1. [Google Scholar] [CrossRef] [PubMed]
- Twarock, R.; Luque, A. Structural Puzzles in Virology Solved with an Overarching Icosahedral Design Principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef]
- Dykeman, E.C.; Stockley, P.G.; Twarock, R. Solving a Levinthal’s Paradox for Virus Assembly Identifies a Unique Antiviral Strategy. Proc. Natl. Acad. Sci. USA 2014, 111, 5361–5366. [Google Scholar] [CrossRef]
- Garmann, R.F.; Comas-Garcia, M.; Knobler, C.M.; Gelbart, W.M. Physical Principles in the Self-Assembly of a Simple Spherical Virus. Acc. Chem. Res. 2016, 49, 48–55. [Google Scholar] [CrossRef]
- Garmann, R.F.; Goldfain, A.M.; Manoharan, V.N. Measurements of the Self-Assembly Kinetics of Individual Viral Capsids Around Their RNA Genome. Proc. Natl. Acad. Sci. USA 2019, 116, 22485–22490. [Google Scholar] [CrossRef]
- Chevreuil, M.; Law-Hine, D.; Chen, J.; Bressanelli, S.; Combet, S.; Constantin, D.; Degrouard, J.; Möller, J.; Zeghal, M.; Tresset, G. Nonequilibrium Self-Assembly Dynamics of Icosahedral Viral Capsids Packaging Genome or Polyelectrolyte. Nat. Commun. 2018, 9, 3071. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Dragnea, B.; Travesset, A.; Podgornik, R. On virus growth and form. Phys. Rep. 2020, 847, 1–102. [Google Scholar] [CrossRef]
- Comas-Garcia, M.; Garmann, R.F.; Singaram, S.W.; Ben-Shaul, A.; Knobler, C.M.; Gelbart, W.M. Characterization of viral capsid protein self-assembly around short single-stranded RNA. J. Phys. Chem. B 2014, 118, 7510–7519. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; He, S.; Chen, X.; Huang, Z.; Zhou, Z.; Zhou, Z.; Chen, Q.; Chen, S.; Kang, S. Structural Insight Into the SARS-CoV-2 Nucleocapsid Protein C-Terminal Domain Reveals a Novel Recognition Mechanism for Viral Transcriptional Regulatory Sequences. Front. Chem. 2021, 8, 1–12. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef]
- Zinzula, L.; Basquin, J.; Bohn, S.; Beck, F.; Klumpe, S.; Pfeifer, G.; Nagy, I.; Bracher, A.; Hartl, F.U.; Baumeister, W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid-19 severe acute respiratory syndrome coronavirus 2. Biochem. Biophys. Res. Commun. 2021, 538, 54–62. [Google Scholar] [CrossRef]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef]
- Cao, C.; Cai, Z.; Xiao, X.; Rao, J.; Chen, J.; Hu, N.; Yang, M.; Xing, X.; Wang, Y.; Li, M.; et al. The architecture of the SARS-CoV-2 RNA genome inside virion. Nat. Commun. 2021, 12, 3917. [Google Scholar] [CrossRef] [PubMed]
- Lipferta, J.; Skinnera, G.M.; Keegstraa, J.M.; Hensgensa, T.; Jagera, T.; Dulina, D.; Köber, M.; Yu, Z.; Donkers, S.P.; Chou, F.C.; et al. Double-stranded RNA under force and torque: Similarities to and striking differences from double-stranded DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Arias-Gonzalez, J.R. Single-molecule portrait of DNA and RNA double helices. Integr. Biol. 2014, 6, 904–925. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Erdemci-Tandogan, G.; Wagner, J.; Van Der Schoot, P.; Zandi, R. Impact of a nonuniform charge distribution on virus assembly. Phys. Rev. E 2017, 96, 022401. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Bruinsma, R.F. RNA condensation and the wetting transition. Phys. Rev. Lett. 2006, 97, 108102. [Google Scholar] [CrossRef]
- Manning, G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978, 11, 179–246. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glaser, J.; Glotzer, S.C. HOOMD-blue: A Python package for high-performance molecular dynamics and hard particle Monte Carlo simulations. Comput. Mater. Sci. 2020, 173, 109363. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Modelling Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Panahandeh, S.; Li, S.; Zandi, R. The Equilibrium Structure of Self-Assembled Protein Nano-Cages. Nanoscale 2018, 10, 22802–22809. [Google Scholar] [CrossRef] [PubMed]
- Belyi, V.A.; Muthukumar, M. Electrostatic origin of the genome packing in viruses. Proc. Natl. Acad. Sci. USA 2006, 103, 17174–17178. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, R.; Wielstra, P.; Calcines-Cruz, C.; van Waveren, T.; Hernandez-Garcia, A.; van der Schoot, P. A nucleation-and-growth model for the packaging of genome in linear virus-like particles: Impact of multiple packaging signals. bioRxiv 2022. [Google Scholar] [CrossRef]
- Comas-Garcia, M.; Cadena-Nava, R.D.; Rao, A.L.N.; Knobler, C.M.; Gelbart, W.M. In Vitro Quantification of the Relative Packaging Efficiencies of Single-Stranded RNA Molecules by Viral Capsid Protein. J. Virol. 2012, 86, 12271–12282. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A Severe Acute Respiratory Syndrome Coronavirus That Lacks the E Gene Is Attenuated In Vitro and In Vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 Virus-Like Particles by Mammalian Expression System. Front. Bioeng. Biotechnol. 2020, 8, 1–6. [Google Scholar] [CrossRef]
- Thomas, S. The structure of the membrane protein of sars-cov-2 resembles the sugar transporter semisweet. Pathog. Immun. 2020, 5, 342–363. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Modeling of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Proteins by Machine Learning and Physics-Based Refinement. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dolan, K.A.; Dutta, M.; Kern, D.M.; Kotecha, A.; Voth, G.A.; Brohawn, S.G. Structure of SARS-CoV-2 M protein in lipid nanodiscs. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kuo, L.; Koetzner, C.A.; Masters, P.S. A key role for the carboxy-terminal tail of the murine coronavirus nucleocapsid protein in coordination of genome packaging. Virology 2016, 494, 100–107. [Google Scholar] [CrossRef]
- Verma, S.; Bednar, V.; Blount, A.; Hogue, B.G. Identification of Functionally Important Negatively Charged Residues in the Carboxy End of Mouse Hepatitis Coronavirus A59 Nucleocapsid Protein. J. Virol. 2006, 80, 4344–4355. [Google Scholar] [CrossRef]
- Luo, H.; Wu, D.; Shen, C.; Chen, K.; Shen, X.; Jiang, H. Severe acute respiratory syndrome coronavirus membrane protein interacts with nucleocapsid protein mostly through their carboxyl termini by electrostatic attraction. Int. J. Biochem. Cell Biol. 2006, 38, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Siber, A.; Bozic, A.L.; Podgornik, R. Energies and pressures in viruses: Contribution of nonspecific electrostatic interactions. Phys. Chem. Chem. Phys. 2012, 14, 3746. [Google Scholar] [CrossRef]
- Savastano, A.; Ibáñez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for its Dual Functions. Mol. Cell 2020, 80, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H.; et al. Targeting liquid–liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Cai, S.; Zhuang, Z.; Zhao, Z.; Jin, S.; Xie, W.; Zhou, L.; Zhang, L.; Zhao, J.; et al. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-κB hyper-activation and inflammation. Signal Transduct. Target. Ther. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Scherer, K.M.; Mascheroni, L.; Carnell, G.W.; Wunderlich, L.C.; Makarchuk, S.; Brockhoff, M.; Mela, I.; Fernandez-Villegas, A.; Barysevich, M.; Stewart, H.; et al. SARS-CoV-2 nucleocapsid protein adheres to replication organelles before viral assembly at the Golgi/ERGIC and lysosome-mediated egress. Sci. Adv. 2022, 8, eabl4895. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zandi, R. Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding. Viruses 2022, 14, 2089. https://doi.org/10.3390/v14102089
Li S, Zandi R. Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding. Viruses. 2022; 14(10):2089. https://doi.org/10.3390/v14102089
Chicago/Turabian StyleLi, Siyu, and Roya Zandi. 2022. "Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding" Viruses 14, no. 10: 2089. https://doi.org/10.3390/v14102089
APA StyleLi, S., & Zandi, R. (2022). Biophysical Modeling of SARS-CoV-2 Assembly: Genome Condensation and Budding. Viruses, 14(10), 2089. https://doi.org/10.3390/v14102089