
A Recombinant System and Reporter Viruses for Papiine Alphaherpesvirus 2

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids, Oligonucleotides and Microorganisms
2.2. Cells and Virus
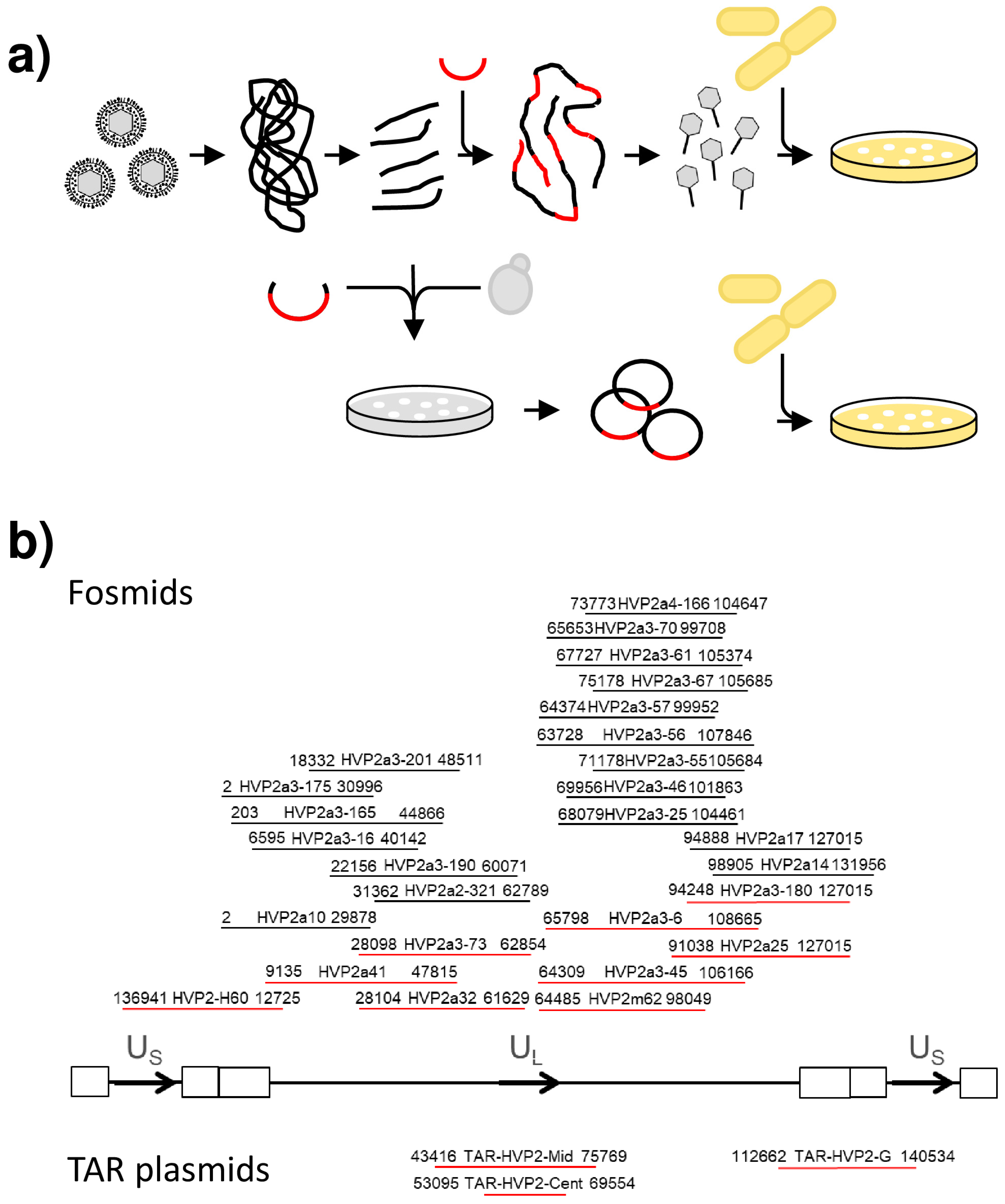
2.3. Fosmid Cloning
2.4. Transformation-Associated Recombination
2.5. Recombineering
2.6. Virus Rescue
2.7. High-Throughput Sequencing and De Novo Assembly
2.8. Plaque Assay and Virus Growth Curves
2.9. Microscopy
2.10. Infection and Luciferase Assay
2.11. Inhibitor Assay
2.12. Neutralization Assay
2.13. Statistical Analysis
3. Results
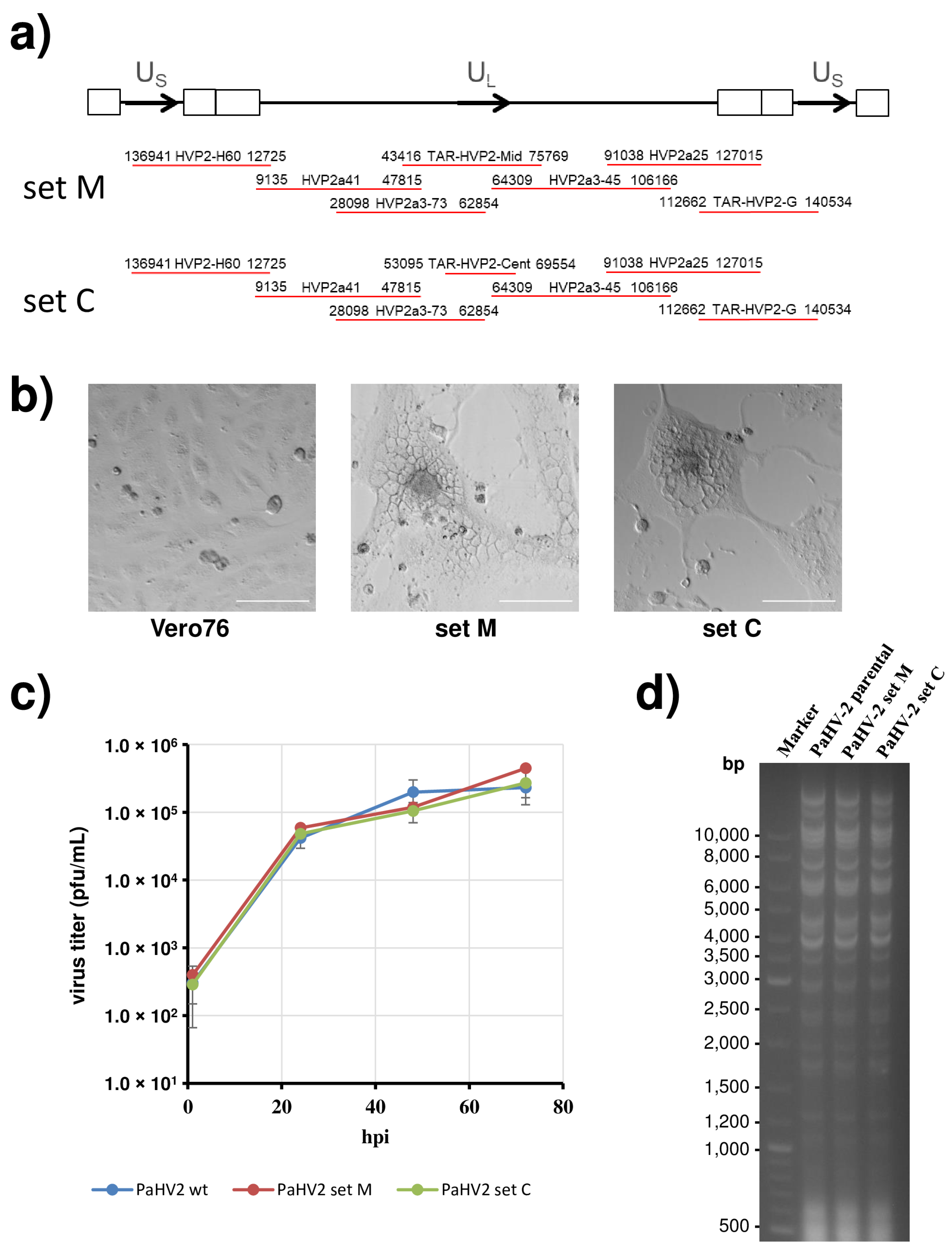
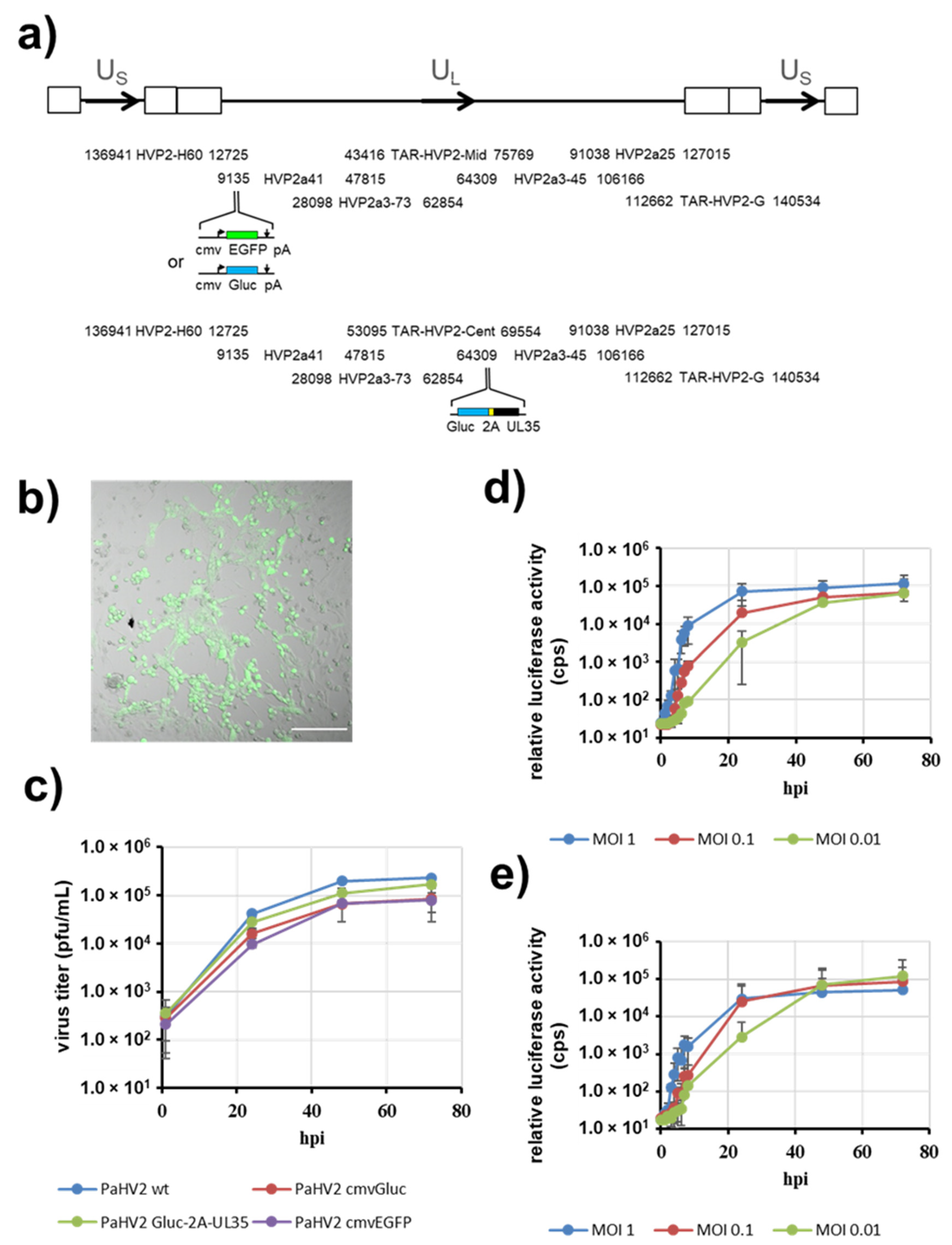
3.1. Cloning of the Papiine Alphaherpesvirus 2 Genome
3.2. PaHV2 Genomes with Reporter Genes
3.3. Inhibition of Reporter Virus Infection by Antibodies and Antivirals
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Azab, W.; Dayaram, A.; Greenwood, A.D.; Osterrieder, N. How Host Specific Are Herpesviruses? Lessons from Herpesviruses Infecting Wild and Endangered Mammals. Annu. Rev. Virol. 2018, 5, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Voevodin, A.F.; Marx, P.A. Simplexviruses. In Simian Virology; Wiley-Blackwell: Ames, IA, USA, 2009; pp. 267–293. [Google Scholar]
- Kolb, A.W.; Brandt, C.R. Genomic nucleotide-based distance analysis for delimiting old world monkey derived herpes simplex virus species. BMC Genom. 2020, 21, 436. [Google Scholar] [CrossRef]
- Tyler, S.D.; Severini, A. The complete genome sequence of herpesvirus papio 2 (Cercopithecine herpesvirus 16) shows evidence of recombination events among various progenitor herpesviruses. J. Virol. 2006, 80, 1214–1221. [Google Scholar] [CrossRef][Green Version]
- Eberle, R.; Black, D.H.; Blewett, E.L.; White, G.L. Prevalence of Herpesvirus papio 2 in baboons and identification of immunogenic viral polypeptides. Lab. Anim. Sci. 1997, 47, 256–262. [Google Scholar] [PubMed]
- Ritchey, J.W.; Ealey, K.A.; Payton, M.E.; Eberle, R. Comparative pathology of infections with baboon and African green monkey alpha-herpesviruses in mice. J. Comp. Pathol. 2002, 127, 150–161. [Google Scholar] [CrossRef]
- Brush, L.A.; Black, D.H.; McCormack, K.A.; Maxwell, L.K.; Wright, G.; Ritchey, J.W.; Payton, M.E.; Eberle, R. Papiine herpesvirus 2 as a predictive model for drug sensitivity of Macacine herpesvirus 1 (monkey B virus). Comp. Med. 2014, 64, 386–393. [Google Scholar]
- Rogers, K.M.; Ealey, K.A.; Ritchey, J.W.; Black, D.H.; Eberle, R. Pathogenicity of different baboon herpesvirus papio 2 isolates is characterized by either extreme neurovirulence or complete apathogenicity. J. Virol. 2003, 77, 10731–10739. [Google Scholar] [CrossRef][Green Version]
- Black, D.; Ohsawa, K.; Tyler, S.; Maxwell, L.; Eberle, R. A single viral gene determines lethal cross-species neurovirulence of baboon herpesvirus HVP2. Virology 2014, 452, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Davison, A.J. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 1993, 197, 116–124. [Google Scholar] [CrossRef]
- Chukhno, E.; Gartner, S.; Rahman Siregar, A.; Mehr, A.; Wende, M.; Petkov, S.; Gotting, J.; Dhingra, A.; Schulz, T.; Pohlmann, S.; et al. A Fosmid-Based System for the Generation of Recombinant Cercopithecine Alphaherpesvirus 2 Encoding Reporter Genes. Viruses 2019, 11, 1026. [Google Scholar] [CrossRef]
- Li, K.; Liu, Y.; Liu, C.; Gao, L.; Zhang, Y.; Cui, H.; Gao, Y.; Qi, X.; Zhong, L.; Wang, X. Recombinant Marek’s disease virus type 1 provides full protection against very virulent Marek’s and infectious bursal disease viruses in chickens. Sci. Rep. 2016, 6, 39263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Abid, M.; Yin, H.; Wu, H.; Teklue, T.; Qiu, H.J.; Sun, Y. Establishment of an Efficient and Flexible Genetic Manipulation Platform Based on a Fosmid Library for Rapid Generation of Recombinant Pseudorabies Virus. Front. Microbiol. 2018, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Messerle, M.; Crnkovic, I.; Hammerschmidt, W.; Ziegler, H.; Koszinowski, U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 1997, 94, 14759–14763. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kagawa, H.; Yamanashi, Y.; Sata, T.; Kawaguchi, Y. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: Viruses reconstituted from the clone exhibit wild-type properties In Vitro and In Vivo. J. Virol. 2003, 77, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Meseda, C.A.; Schmeisser, F.; Pedersen, R.; Woerner, A.; Weir, J.P. DNA immunization with a herpes simplex virus 2 bacterial artificial chromosome. Virology 2004, 318, 420–428. [Google Scholar] [CrossRef]
- Oldfield, L.M.; Grzesik, P.; Voorhies, A.A.; Alperovich, N.; MacMath, D.; Najera, C.D.; Chandra, D.S.; Prasad, S.; Noskov, V.N.; Montague, M.G.; et al. Genome-wide engineering of an infectious clone of herpes simplex virus type 1 using synthetic genomics assembly methods. Proc. Natl. Acad. Sci. USA 2017, 114, E8885–E8894. [Google Scholar] [CrossRef] [PubMed]
- Vashee, S.; Stockwell, T.B.; Alperovich, N.; Denisova, E.A.; Gibson, D.G.; Cady, K.C.; Miller, K.; Kannan, K.; Malouli, D.; Crawford, L.B.; et al. Cloning, Assembly, and Modification of the Primary Human Cytomegalovirus Isolate Toledo by Yeast-Based Transformation-Associated Recombination. mSphere 2017, 2, e00331-17. [Google Scholar] [CrossRef]
- Thi Nhu Thao, T.; Labroussaa, F.; Ebert, N.; V’Kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef]
- Gietz, R.D.; Sugino, A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 1988, 74, 527–534. [Google Scholar] [CrossRef]
- Grant, S.G.; Jessee, J.; Bloom, F.R.; Hanahan, D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 1990, 87, 4645–4649. [Google Scholar] [CrossRef]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. In In Vitro Mutagenesis Protocols; Humana Press: Totowa, NJ, USA, 2010; Volume 634, pp. 421–430. [Google Scholar] [CrossRef]
- Doherty, J.P.; Lindeman, R.; Trent, R.J.; Graham, M.W.; Woodcock, D.M. Escherichia coli host strains SURE and SRB fail to preserve a palindrome cloned in lambda phage: Improved alternate host strains. Gene 1993, 124, 29–35. [Google Scholar] [CrossRef]
- Noskov, V.N.; Kouprina, N.; Leem, S.H.; Ouspenski, I.; Barrett, J.C.; Larionov, V. A general cloning system to selectively isolate any eukaryotic or prokaryotic genomic region in yeast. BMC Genom. 2003, 4, 16. [Google Scholar] [CrossRef]
- Chang, W.L.; Kirchoff, V.; Pari, G.S.; Barry, P.A. Replication of rhesus cytomegalovirus in life-expanded rhesus fibroblasts expressing human telomerase. J. Virol. Methods 2002, 104, 135–146. [Google Scholar] [CrossRef]
- Kuo, L.; Godeke, G.J.; Raamsman, M.J.; Masters, P.S.; Rottier, P.J. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: Crossing the host cell species barrier. J. Virol. 2000, 74, 1393–1406. [Google Scholar] [CrossRef]
- Dirks, W.G.; Drexler, H.G. STR DNA typing of human cell lines: Detection of intra- and interspecies cross-contamination. In Basic Cell Culture Protocols; Humana Press: Totowa, NJ, USA, 2013; Volume 946, pp. 27–38. [Google Scholar] [CrossRef]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Paabo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196–6200. [Google Scholar] [CrossRef]
- Kouprina, N.; Larionov, V. Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 2008, 3, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Noskov, V.N.; Larionov, V. Selective isolation of large segments from individual microbial genomes and environmental DNA samples using transformation-associated recombination cloning in yeast. Nat. Protoc. 2020, 15, 734–749. [Google Scholar] [CrossRef]
- Thapana, W.; Sujiwattanarat, P.; Srikulnath, K.; Hirai, H.; Koga, A. Reduction in the structural instability of cloned eukaryotic tandem-repeat DNA by low-temperature culturing of host bacteria. Genet. Res. 2014, 96, e13. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Sauerbrei, A.; Bohn, K. Phenotypic and genotypic testing of HSV-1 resistance to antivirals. Methods Mol. Biol. 2014, 1144, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Pauls, F.P.; Dowdle, W.R. A serologic study of herpesvirus hominis strains by microneutralization tests. J. Immunol. 1967, 98, 941–947. [Google Scholar] [PubMed]
- Rawls, W.E.; Iwamoto, K.; Adam, E.; Melnick, J.L. Measurement of antibodies to herpesvirus types 1 and 2 in human sera. J. Immunol. 1970, 104, 599–606. [Google Scholar]
- Stalder, H.; Oxman, M.N.; Herrmann, K.L. Herpes simplex virus microneutralization: A simplification of the test. J. Infect. Dis. 1975, 131, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Gauger, P.C.; Vincent, A.L. Serum virus neutralization assay for detection and quantitation of serum-neutralizing antibodies to influenza A virus in swine. Methods Mol. Biol. 2014, 1161, 313–324. [Google Scholar] [CrossRef]
- Kaul, A.; Schonmann, U.; Pohlmann, S. Seroprevalence of viral infections in captive rhesus and cynomolgus macaques. Primate Biol. 2019, 6, 1–6. [Google Scholar] [CrossRef]
- Ferrara, F.; Temperton, N. Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis. Methods Protoc. 2018, 1, 8. [Google Scholar] [CrossRef]
- Morimoto, T.; Arii, J.; Akashi, H.; Kawaguchi, Y. Identification of multiple sites suitable for insertion of foreign genes in herpes simplex virus genomes. Microbiol. Immunol. 2009, 53, 155–161. [Google Scholar] [CrossRef]
- Ito, K.; Chiba, S. Arrest peptides: Cis-acting modulators of translation. Annu. Rev. Biochem. 2013, 82, 171–202. [Google Scholar] [CrossRef]
- Eckert, N.; Wrensch, F.; Gartner, S.; Palanisamy, N.; Goedecke, U.; Jager, N.; Pohlmann, S.; Winkler, M. Influenza A virus encoding secreted Gaussia luciferase as useful tool to analyze viral replication and its inhibition by antiviral compounds and cellular proteins. PLoS ONE 2014, 9, e97695. [Google Scholar] [CrossRef]
- Monier, K.; Armas, J.C.; Etteldorf, S.; Ghazal, P.; Sullivan, K.F. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and In Vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Verkhusha, V.V.; Kuznetsova, I.M.; Stepanenko, O.V.; Zaraisky, A.G.; Shavlovsky, M.M.; Turoverov, K.K.; Uversky, V.N. High stability of Discosoma DsRed as compared to Aequorea EGFP. Biochemistry 2003, 42, 7879–7884. [Google Scholar] [CrossRef]
- Wurdinger, T.; Badr, C.; Pike, L.; de Kleine, R.; Weissleder, R.; Breakefield, X.O.; Tannous, B.A. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 2008, 5, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Payton, M.E.; d’Offay, J.M.; Prado, M.E.; Black, D.H.; Damania, B.; White, G.L.; Eberle, R. Comparative transmission of multiple herpesviruses and simian virus 40 in a baboon breeding colony. Comp. Med. 2004, 54, 695–704. [Google Scholar]
- Eberle, R.; Black, D.H.; Lehenbauer, T.W.; White, G.L. Shedding and transmission of baboon Herpesvirus papio 2 (HVP2) in a breeding colony. Lab. Anim. Sci. 1998, 48, 23–28. [Google Scholar] [PubMed]
- Kalter, S.S.; Hilliard, J.K.; Heberling, R.L. The differential diagnosis of herpesvirus infections in man and animals. Dev. Biol. Stand. 1982, 52, 101–113. [Google Scholar]
- Hampar, B.; Martos, L.M.; Chakrabarty, M.; Burroughs, M.A. Late 19S rabbit antibody neutralization test for differentiating herpes simplex virus types 1 and 2. J. Immunol. 1970, 104, 593–598. [Google Scholar]
- Katz, D.; Shi, W.; Krug, P.W.; Henkel, R.; McClure, H.; Hilliard, J.K. Antibody cross-reactivity of alphaherpesviruses as mirrored in naturally infected primates. Arch. Virol. 2002, 147, 929–941. [Google Scholar] [CrossRef]
- Pohlmann, S.; Kruger, A.; Hafezi, W.; Schneider, S.; Gruber, J.; Winkler, M.; Kaul, A. Detection systems for antibody responses against herpes B virus. Primate Biol. 2017, 4, 9–16. [Google Scholar] [CrossRef]
- Davis, K.L.; Korom, M.; Morrison, L.A. Herpes simplex virus 2 ICP34.5 confers neurovirulence by regulating the type I interferon response. Virology 2014, 468–470, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Luebcke, E.; Dubovi, E.; Black, D.; Ohsawa, K.; Eberle, R. Isolation and characterization of a chimpanzee alphaherpesvirus. J. Gen. Virol. 2006, 87, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.D.; Peters, G.A.; Severini, A. Complete genome sequence of cercopithecine herpesvirus 2 (SA8) and comparison with other simplexviruses. Virology 2005, 331, 429–440. [Google Scholar] [CrossRef]
- Perelygina, L.; Zhu, L.; Zurkuhlen, H.; Mills, R.; Borodovsky, M.; Hilliard, J.K. Complete sequence and comparative analysis of the genome of herpes B virus (Cercopithecine herpesvirus 1) from a rhesus monkey. J. Virol. 2003, 77, 6167–6177. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide Name | Oligonucleotide Sequence (Given in 5′-3′ Direction) |
|---|---|
| Oligonucleotides used for cloning | |
| mcs-FOS-f | AATTCCTCGAGGCTAGCTTAATTAAGGATCCCACGTGGGATCCATCGATACGCGTCGTACGGCATG |
| mcs-FOS-r | CCGTACGACGCGTATCGATGGATCCCACGTGGGATCCTTAATTAAGCTAGCCTCGAGG |
| Gluc-5A | CCGGTACCATGGGAGTCAAAGTTCTG |
| Gluc-3StopN | CCGCGGCCGCTTAGTCACCACCGGCCCCCTTG |
| Oligonucleotides used for TAR | |
| TAR-HVP2-Mid-F | GTAGTGCGCGTCCGCCACCAGCCCCAGCACCGTGTTGGTCGCCGTCTGGAGATCCTC-TAGAGTCGACCTGCAG |
| TAR-HVP2-Mid-R | GCGGTCTATGTGCTTCACCTGCACGAACTCGCTCACGGTGGTGCGCTTGGGTTTAAACGTCGTGACTGGGAAAACCCTG |
| TAR-HVP2-Cent-F | CAGCTCGCCGCAGAGCGACTCGTTAAGAGCCAGGAGGTCGGGGTCGAAGGATCCTC-TAGAGTCGACCTGCAG |
| TAR-HVP2-Cent-R | GCGACACCACCGCCGATCGACCCGCTGTGGAAACCACACGCACATAGACGTTTAAA-CGTCGTGACTGGGAAAACCCTG |
| TAR-HVP2-G-F | GGCTCGCGTTCGTGATCATCACCGTCATGGTGCTGCGGCCATGCCGTCCGATCCTCTAGAGTCGACCTGCAG |
| TAR-HVP2-G-R | CCGTGCTGGCGATCAGCCCCTGGCTCGCCACGCGCAGCAGGATCTCGCAGTTTAAACGTCGTGACTGGGAAAACCCTG |
| Oligonucleotides used for recombineering | |
| ep-HVPUL4pA-cmv | TCCCGCGGCCCCGGGCGCTCCAGAGACGCGGCGAGACGAATAAACGCGGTGTTGACATTGATTATTGACT |
| ep-HVPUL3-gGHpA | CGCCTCGCCGATCCCAAGGATGGATGACACACGAATAAATATTTCAAACGTCCCCAGCATGCCTGCTATT |
| ep-Pa35-Gluc-5 | TCCGCCGCCCTCCTCCCGGTCGCCGTTGCGCCCCGCCCGCCGCCCGCGCCATGGGAGTCAAAGTTCTGTTT |
| ep-Pa35-2A-3 | TCGGGGTGAAGCGTGTCGGGCCGGTGGAACTGCGGGACCGCGGCGGACATCGGGCCCGGGTTTTCTTCCAC |
| Virus Construct | Fosmid | Template | Primer |
|---|---|---|---|
| HVP2-cmvGluc | HVP2a41 | pcDNA3-Gluc-en | ep-HVPUL4pA-cmv ep-HVPUL3-gGHpA |
| HVP2-cmvEGFP | HVP2a41 | pcDNA3-EGFP-en | ep-HVPUL4pA-cmv ep-HVPUL3-gGHpA |
| HVP2-Gluc-2A-UL35 | HVP2a3-45 | pHW2000GG-seg8-A/PR/8/34-M2A-Gluc-en | ep-Pa35-Gluc-5 ep-Pa35-2A-3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman Siregar, A.; Gärtner, S.; Götting, J.; Stegen, P.; Kaul, A.; Schulz, T.F.; Pöhlmann, S.; Winkler, M. A Recombinant System and Reporter Viruses for Papiine Alphaherpesvirus 2. Viruses 2022, 14, 91. https://doi.org/10.3390/v14010091
Rahman Siregar A, Gärtner S, Götting J, Stegen P, Kaul A, Schulz TF, Pöhlmann S, Winkler M. A Recombinant System and Reporter Viruses for Papiine Alphaherpesvirus 2. Viruses. 2022; 14(1):91. https://doi.org/10.3390/v14010091
Chicago/Turabian StyleRahman Siregar, Abdul, Sabine Gärtner, Jasper Götting, Philipp Stegen, Artur Kaul, Thomas F. Schulz, Stefan Pöhlmann, and Michael Winkler. 2022. "A Recombinant System and Reporter Viruses for Papiine Alphaherpesvirus 2" Viruses 14, no. 1: 91. https://doi.org/10.3390/v14010091
APA StyleRahman Siregar, A., Gärtner, S., Götting, J., Stegen, P., Kaul, A., Schulz, T. F., Pöhlmann, S., & Winkler, M. (2022). A Recombinant System and Reporter Viruses for Papiine Alphaherpesvirus 2. Viruses, 14(1), 91. https://doi.org/10.3390/v14010091