
The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2

 ,
,  , ,
, , {kind=link}
Abstract
:1. Introduction
2. Notable Coronaviruses
3. Classification
4. Distribution
5. Genome Architecture
6. Evolution by Point Mutations
7. Evolution by Insertions/Deletions
8. The Progenitor of SARS-CoV-2 Was Already a Generalist Virus That Did Not Need Many Mutations to Adapt to Its Human Hosts
9. Evolution by Recombination
10. Evolution by Intratypic Homologous Recombination
11. Evolution by Intertypic Homologous Recombination
12. Intertypic Recombination Is Modular
13. Evolution by Non-Homologous Recombination among CoVs and with Other Taxa
14. Implications for Vaccine Design and Development
15. Implications for Drug Design and Development
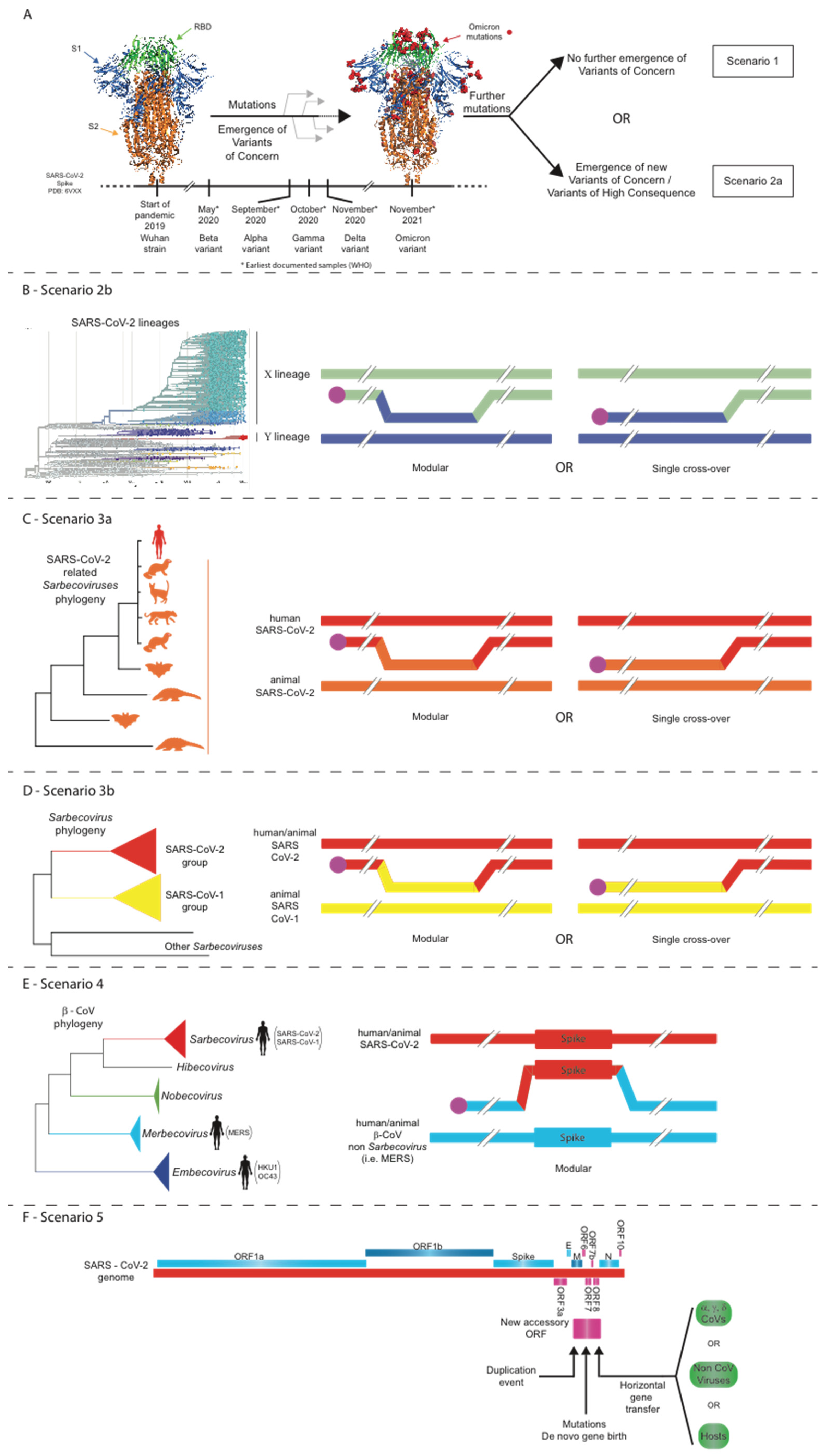
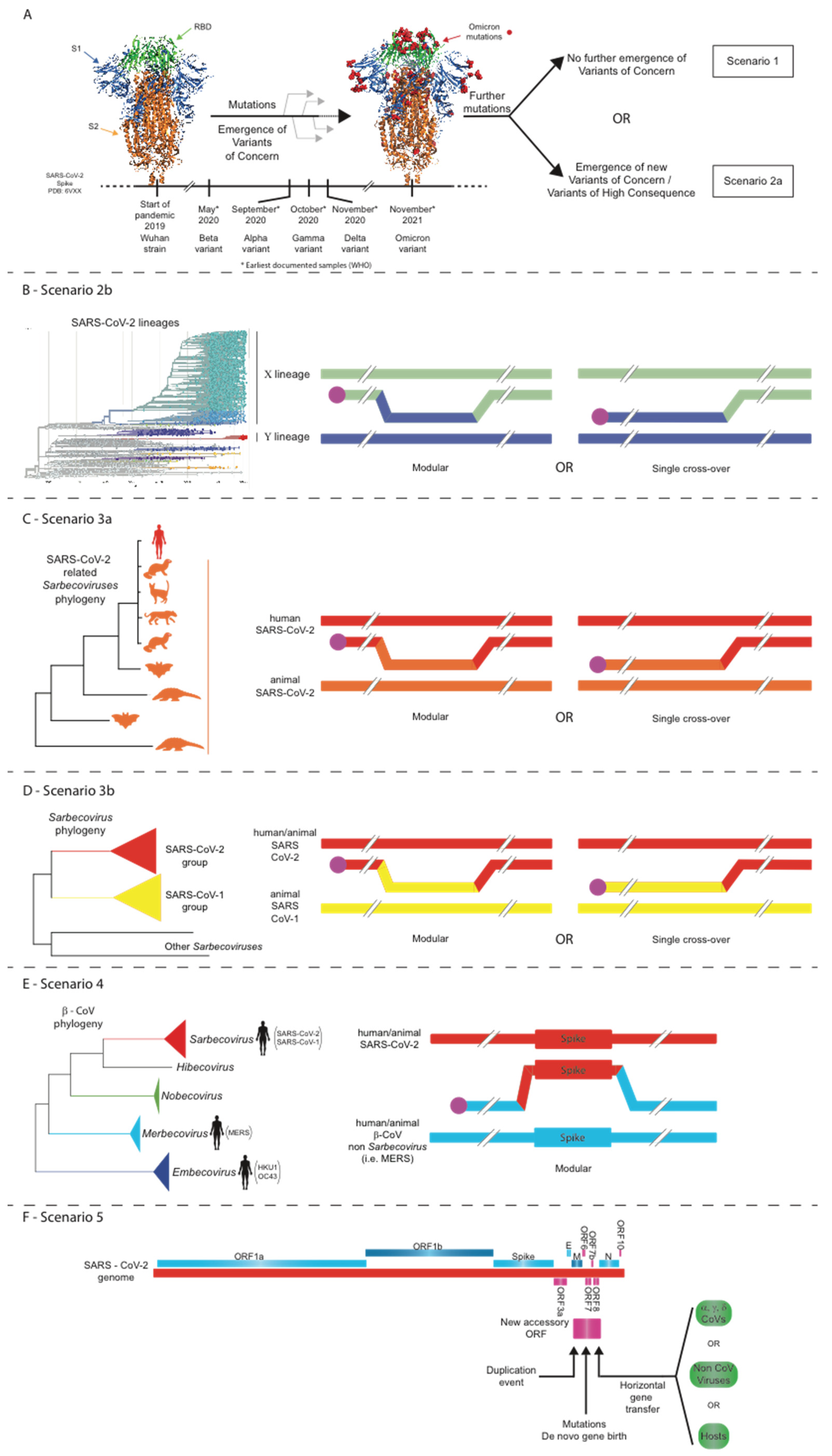
16. Five Scenarios for the Future Evolution of SARS-CoV-2 during the COVID-19 Pandemic
16.1. Scenario 1: Structural Constraints Limit Any Further Evolution of the SARS-CoV-2 Spike
16.2. Scenario 2: Point Mutations, Insertions/Deletions, and/or Intra-SARS-CoV-2 Recombination Events Lead to the Evolution of Novel SARS-CoV-2 Strains
16.3. Scenario 3: Intratypic Recombinations between SARS-CoV-2 and Other Sarbecoviruses
16.4. Scenario 4: Intertypic Recombination between SARS-CoV-2 and Viruses from Other Beta-CoV Subgenera
16.5. Scenario 5: Accessory ORF Acquisition by Non-Homologous Recombination of SARS-CoV-2 with Other Coronaviruses or Even Other Viruses/Hosts or Even via De Novo Gene Birth
17. Conclusions—Implications for Vaccination Policies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3CLpro | 3-chymotrypsin-like protease |
| ACE2 | angiotensin-converting enzyme 2 |
| AOF | accessory ORF family |
| APN | aminopeptidase N |
| CCoV | canine coronavirus |
| CDC | Centers for Disease Control and Prevention |
| CoV | coronavirus |
| COVID-19 | coronavirus disease 2019 |
| DPP4 | dipeptidyl peptidase 4 |
| FCoV | feline coronavirus |
| FCS | furin cleavage site |
| FIPV | feline infectious peritonitis virus |
| GARD | genetic algorithm for recombination detection |
| GISAID | Global Initiative on Sharing All Influenza Data |
| HEL | NSP 13 C-terminal nucleoside triphosphate-binding/helicase motif |
| HIV | human immunodeficiency virus |
| IBV | infectious bronchitis virus |
| mABs | monoclonal antibodies |
| MERS | Middle East respiratory syndrome |
| MHV | murine hepatitis virus |
| N | nucleocapsid |
| NiRAN | nidovirus RdRp-associated nucleotidyl transferase domain |
| NSP | non-structural peptide |
| nt | nucleotide |
| ORF | open reading frame |
| PDCoV | porcine delta coronavirus |
| PEDV | porcine epidemic diarrhea virus |
| RBD | receptor-binding domain |
| RBM | receptor-binding motif within the RBD of spike |
| RDP4 | recombination detection program 4 |
| RdRp | RNA-dependent RNA polymerase |
| SADS | swine acute diarrhea syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| TGEV | transmissible gastroenteritis virus |
| TRS | transcriptional regulatory sequence |
| UTR | untranslated region |
| VoC | variant of concern |
| WHO | World Health Organization |
| ZBD | NSP 13 N-terminal cysteine-rich zinc-binding domain. |
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C.; et al. The Origins of SARS-CoV-2: A Critical Review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Wang, M.; Lau, S.K.P.; Xu, H.; Poon, R.W.S.; Guo, R.; Wong, B.H.L.; Gao, K.; Tsoi, H.-W.; Huang, Y.; et al. Comparative Analysis of Twelve Genomes of Three Novel Group 2c and Group 2d Coronaviruses Reveals Unique Group and Subgroup Features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-H.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Bermingham, A.; Chand, M.A.; Brown, C.S.; Aarons, E.; Tong, C.; Langrish, C.; Hoschler, K.; Brown, K.; Galiano, M.; Myers, R.; et al. Severe Respiratory Illness Caused by a Novel Coronavirus, in a Patient Transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill. 2012, 17, 20290. [Google Scholar] [CrossRef]
- Pascual-Iglesias, A.; Sanchez, C.M.; Penzes, Z.; Sola, I.; Enjuanes, L.; Zuñiga, S. Recombinant Chimeric Transmissible Gastroenteritis Virus (TGEV)—Porcine Epidemic Diarrhea Virus (PEDV) Virus Provides Protection against Virulent PEDV. Viruses 2019, 11, 682. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.R.; Navas-Martin, S. Coronavirus Pathogenesis and the Emerging Pathogen Severe Acute Respiratory Syndrome Coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the Largest RNA Virus Genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Saberi, A.; Gulyaeva, A.A.; Brubacher, J.L.; Newmark, P.A.; Gorbalenya, A.E. A Planarian Nidovirus Expands the Limits of RNA Genome Size. PLoS Pathog. 2018, 14, e1007314. [Google Scholar] [CrossRef] [Green Version]
- Gulyaeva, A.A.; Gorbalenya, A.E. A Nidovirus Perspective on SARS-CoV-2. Biochem. Biophys. Res. Commun. 2021, 538, 24–34. [Google Scholar] [CrossRef]
- ICTV Coronaviridae Study Group International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/223/coronaviridae-figures (accessed on 17 September 2020).
- Holmes, K.V. Coronaviruses (Coronaviridae). Encycl. Virol. 1999, 291–298. [Google Scholar] [CrossRef]
- Lauber, C.; Ziebuhr, J.; Junglen, S.; Drosten, C.; Zirkel, F.; Nga, P.T.; Morita, K.; Snijder, E.J.; Gorbalenya, A.E. Mesoniviridae: A Proposed New Family in the Order Nidovirales Formed by a Single Species of Mosquito-Borne Viruses. Arch. Virol. 2012, 157, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.; Gorbalenya, A.E. Partitioning the Genetic Diversity of a Virus Family: Approach and Evaluation through a Case Study of Picornaviruses. J. Virol. 2012, 86, 3890–3904. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern Naming Scheme Conducive for Global Discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary Trajectory of SARS-CoV-2 and Emerging Variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 2 November 2021).
- Ferguson, N.; Ghani, A.; Cori, A.; Hogan, A.; Hinsley, W.; Volz, E. Report 49: Growth, Population Distribution and Immune Escape of Omicron in England; Imperial College London: London, UK, 2021. [Google Scholar] [CrossRef]
- Cameroni, E.; Saliba, C.; Bowen, J.E.; Rosen, L.E.; Culap, K.; Pinto, D.; VanBlargan, L.A.; Marco, A.D.; Zepeda, S.K.; di Iulio, J.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2021. [Google Scholar] [CrossRef]
- FDA. SARS-CoV-2 Viral Mutations: Impact on COVID-19 Tests; FDA: Silver Spring, MD, USA, 2021. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests (accessed on 23 November 2021).
- Wille, M.; Holmes, E.C. Wild Birds as Reservoirs for Diverse and Abundant Gamma- and Deltacoronaviruses. FEMS Microbiol. Rev. 2020, 44, 631–644. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, L.; Liu, P.; Li, H.; Huo, S.; Zong, K.; Zhu, S.; Guo, Y.; Zhang, L.; Hu, B.; et al. A Novel Potentially Recombinant Rodent Coronavirus with a Polybasic Cleavage Site in the Spike Protein. J. Virol. 2021, 95, e0117321. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Lau, S.K.P.; Woo, P.C.Y. Interspecies Jumping of Bat Coronaviruses. Viruses 2021, 13, 2188. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, K.; Shi, Z.-L.; Zhou, P. Bat Coronaviruses in China. Viruses 2019, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Song, H.-D.; Tu, C.-C.; Zhang, G.-W.; Wang, S.-Y.; Zheng, K.; Lei, L.-C.; Chen, Q.-X.; Gao, Y.-W.; Zhou, H.-Q.; Xiang, H.; et al. Cross-Host Evolution of Severe Acute Respiratory Syndrome Coronavirus in Palm Civet and Human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar] [CrossRef] [Green Version]
- Reusken, C.B.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.-J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.-D.; Corman, V.M.; et al. Middle East Respiratory Syndrome Coronavirus Neutralising Serum Antibodies in Dromedary Camels: A Comparative Serological Study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.-J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-Related Coronavirus from Malayan Pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Emergence of Porcine Delta-Coronavirus Pathogenic Infections among Children in Haiti through Independent Zoonoses and Convergent Evolution. medRxiv 2021. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Diaz, A.; Damtie, D.; Xiu, L.; Toh, T.-H.; Lee, J.S.-Y.; Saif, L.J.; Gray, G.C. Novel Canine Coronavirus Isolated from a Hospitalized Pneumonia Patient, East Malaysia. Clin. Infect. Dis. 2021, ciab456. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Hulswit, R.J.G.; Kenney, S.P.; Widjaja, I.; Jung, K.; Alhamo, M.A.; van Dieren, B.; van Kuppeveld, F.J.M.; Saif, L.J.; Bosch, B.-J. Broad Receptor Engagement of an Emerging Global Coronavirus May Potentiate Its Diverse Cross-Species Transmissibility. Proc. Natl. Acad. Sci. USA 2018, 115, E5135–E5143. [Google Scholar] [CrossRef] [Green Version]
- Boley, P.A.; Alhamo, M.A.; Lossie, G.; Yadav, K.K.; Vasquez-Lee, M.; Saif, L.J.; Kenney, S.P. Porcine Deltacoronavirus Infection and Transmission in Poultry, United States. Emerg. Infect. Dis. 2020, 26, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV Poised for Human Emergence. Proc. Natl. Acad. Sci. USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. A SARS-like Cluster of Circulating Bat Coronaviruses Shows Potential for Human Emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef]
- Li, W.; Wong, S.-K.; Li, F.; Kuhn, J.H.; Huang, I.-C.; Choe, H.; Farzan, M. Animal Origins of the Severe Acute Respiratory Syndrome Coronavirus: Insight from ACE2-S-Protein Interactions. J. Virol. 2006, 80, 4211–4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temmam, S.; Vongphayloth, K.; Salazar, E.B.; Munier, S.; Bonomi, M.; Régnault, B.; Douangboubpha, B.; Karami, Y.; Chretien, D.; Sanamxay, D.; et al. Coronaviruses with a SARS-CoV-2-like Receptor-Binding Domain Allowing ACE2-Mediated Entry into Human Cells Isolated from Bats of Indochinese Peninsula. ResearchSquare 2021. [Google Scholar] [CrossRef]
- Olival, K.J.; Cryan, P.M.; Amman, B.R.; Baric, R.S.; Blehert, D.S.; Brook, C.E.; Calisher, C.H.; Castle, K.T.; Coleman, J.T.H.; Daszak, P.; et al. Possibility for Reverse Zoonotic Transmission of SARS-CoV-2 to Free-Ranging Wildlife: A Case Study of Bats. PLoS Pathog. 2020, 16, e1008758. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.V.; Martins, M.; Falkenberg, S.; Buckley, A.; Caserta, L.C.; Mitchell, P.K.; Cassmann, E.D.; Rollins, A.; Zylich, N.C.; Renshaw, R.W.; et al. Susceptibility of White-Tailed Deer (Odocoileus Virginianus) to SARS-CoV-2. J. Virol. 2021, 95, e00083-21. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on Mink Farms between Humans and Mink and Back to Humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef]
- Hoffmann, M.; Zhang, L.; Krüger, N.; Graichen, L.; Kleine-Weber, H.; Hofmann-Winkler, H.; Kempf, A.; Nessler, S.; Riggert, J.; Winkler, M.S.; et al. SARS-CoV-2 Mutations Acquired in Mink Reduce Antibody-Mediated Neutralization. Cell Rep. 2021, 35, 109017. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Non-Homologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2021, msab292. [Google Scholar] [CrossRef]
- Lauber, C.; Goeman, J.J.; Parquet, M.D.C.; Thi Nga, P.; Snijder, E.J.; Morita, K.; Gorbalenya, A.E. The Footprint of Genome Architecture in the Largest Genome Expansion in RNA Viruses. PLoS Pathog. 2013, 9, e1003500. [Google Scholar] [CrossRef] [Green Version]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The Molecular Virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Phillips, J.J.; Chua, M.M.; Lavi, E.; Weiss, S.R. Pathogenesis of Chimeric MHV4/MHV-A59 Recombinant Viruses: The Murine Coronavirus Spike Protein Is a Major Determinant of Neurovirulence. J. Virol. 1999, 73, 7752–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, C.M.; Izeta, A.; Sánchez-Morgado, J.M.; Alonso, S.; Sola, I.; Balasch, M.; Plana-Durán, J.; Enjuanes, L. Targeted Recombination Demonstrates That the Spike Gene of Transmissible Gastroenteritis Coronavirus Is a Determinant of Its Enteric Tropism and Virulence. J. Virol. 1999, 73, 7607–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casais, R.; Dove, B.; Cavanagh, D.; Britton, P. Recombinant Avian Infectious Bronchitis Virus Expressing a Heterologous Spike Gene Demonstrates That the Spike Protein Is a Determinant of Cell Tropism. J. Virol. 2003, 77, 9084–9089. [Google Scholar] [CrossRef] [Green Version]
- Rottier, P.J.M.; Nakamura, K.; Schellen, P.; Volders, H.; Haijema, B.J. Acquisition of Macrophage Tropism during the Pathogenesis of Feline Infectious Peritonitis Is Determined by Mutations in the Feline Coronavirus Spike Protein. J. Virol. 2005, 79, 14122–14130. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.; Godeke, G.J.; Raamsman, M.J.; Masters, P.S.; Rottier, P.J. Retargeting of Coronavirus by Substitution of the Spike Glycoprotein Ectodomain: Crossing the Host Cell Species Barrier. J. Virol. 2000, 74, 1393–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, P.; Fang, L.; Zhang, H.; Xia, S.; Xiao, S. Functions of Coronavirus Accessory Proteins: Overview of the State of the Art. Viruses 2021, 13, 1139. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a Protein of SARS-CoV-2 Induces Apoptosis in Cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-G.; Huang, W.; Lee, H.; van de Leemput, J.; Kane, M.A.; Han, Z. Characterization of SARS-CoV-2 Proteins Reveals Orf6 Pathogenicity, Subcellular Localization, Host Interactions and Attenuation by Selinexor. Cell Biosci. 2021, 11, 58. [Google Scholar] [CrossRef]
- Shang, J.; Han, N.; Chen, Z.; Peng, Y.; Li, L.; Zhou, H.; Ji, C.; Meng, J.; Jiang, T.; Wu, A. Compositional Diversity and Evolutionary Pattern of Coronavirus Accessory Proteins. Brief. Bioinform. 2021, 22, 1267–1278. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High Fidelity of Murine Hepatitis Virus Replication Is Decreased in Nsp14 Exoribonuclease Mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [Green Version]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The Coronavirus Proofreading Exoribonuclease Mediates Extensive Viral Recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA Virus Error Catastrophe: Direct Molecular Test by Using Ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Perales, C. Viral Quasispecies. PLoS Genet 2019, 15, e1008271. [Google Scholar] [CrossRef] [Green Version]
- Borges, V.; Alves, M.J.; Amicone, M.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation Rate of SARS-CoV-2 and Emergence of Mutators during Experimental Evolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Eigen, M. Error Catastrophe and Antiviral Strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An Orally Bioavailable Broad-Spectrum Antiviral Inhibits SARS-CoV-2 in Human Airway Epithelial Cell Cultures and Multiple Coronaviruses in Mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, B.; Campbell, E.A. Molnupiravir: Coding for Catastrophe. Nat. Struct. Mol. Biol. 2021, 28, 706–708. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 Spike D614G Change Enhances Replication and Transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Neches, R.Y.; McGee, M.D.; Kyrpides, N.C. Recombination Should Not Be an Afterthought. Nat. Rev. Microbiol. 2020, 18, 606. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The Emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 Evolution during Treatment of Chronic Infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [Green Version]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. Natural Selection in the Evolution of SARS-CoV-2 in Bats Created a Generalist Virus and Highly Capable Human Pathogen. PLoS Biol. 2021, 19, e3001115. [Google Scholar] [CrossRef]
- Morel, B.; Barbera, P.; Czech, L.; Bettisworth, B.; Hübner, L.; Lutteropp, S.; Serdari, D.; Kostaki, E.-G.; Mamais, I.; Kozlov, A.M.; et al. Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult. Mol. Biol. Evol. 2021, 38, 1777–1791. [Google Scholar] [CrossRef]
- Jaroszewski, L.; Iyer, M.; Alisoltani, A.; Sedova, M.; Godzik, A. The Interplay of SARS-CoV-2 Evolution and Constraints Imposed by the Structure and Functionality of Its Proteins. PLoS Comput. Biol. 2021, 17, e1009147. [Google Scholar] [CrossRef]
- Mei, H.; Kosakovsky Pond, S.; Nekrutenko, A. Stepwise Evolution and Exceptional Conservation of ORF1a/b Overlap in Coronaviruses. Mol. Biol. Evol. 2021, 38, 5678–5684. [Google Scholar] [CrossRef]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Insertions in SARS-CoV-2 Genome Caused by Template Switch and Duplications Give Rise to New Variants of Potential Concern. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, G.; Yu, D.; Cao, R.; Wu, X.; Ling, Y.; Pan, Y.-H.; Yi, C.; Sun, X.; Sun, B.; et al. A Kozak-Related Non-Coding Deletion Effectively Increases B.1.1.7 Transmissibility. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic Determinants of Pathogenicity in SARS-CoV-2 and Other Human Coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; PHE Virology Consortium; de Silva, T.I.; Barclay, W.S. The SARS-CoV-2 Variants Associated with Infections in India, B.1.617, Show Enhanced Spike Cleavage by Furin. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ntountoumi, C.; Vlastaridis, P.; Mossialos, D.; Stathopoulos, C.; Iliopoulos, I.; Promponas, V.; Oliver, S.G.; Amoutzias, G.D. Low Complexity Regions in the Proteins of Prokaryotes Perform Important Functional Roles and Are Highly Conserved. Nucleic Acids Res. 2019, 47, 9998–10009. [Google Scholar] [CrossRef] [PubMed]
- Conceicao, C.; Thakur, N.; Human, S.; Kelly, J.T.; Logan, L.; Bialy, D.; Bhat, S.; Stevenson-Leggett, P.; Zagrajek, A.K.; Hollinghurst, P.; et al. The SARS-CoV-2 Spike Protein Has a Broad Tropism for Mammalian ACE2 Proteins. PLoS Biol. 2020, 18, e3001016. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and Characterization of a Bat SARS-like Coronavirus That Uses the ACE2 Receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Brown, J.; Pedersen, B.S.; Quinlan, A.R.; Elde, N.C. Extensive Recombination-Driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens. bioRxiv 2021. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Wong, E.Y.M.; Tsang, C.-C.; Ahmed, S.S.; Au-Yeung, R.K.H.; Yuen, K.-Y.; Wernery, U.; Woo, P.C.Y. Discovery and Sequence Analysis of Four Deltacoronaviruses from Birds in the Middle East Reveal Interspecies Jumping with Recombination as a Potential Mechanism for Avian-to-Avian and Avian-to-Mammalian Transmission. J. Virol. 2018, 92, e00265-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-Frequency RNA Recombination of Murine Coronaviruses. J. Virol. 1986, 57, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A Contemporary View of Coronavirus Transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Deming, D.J.; Deming, M.E.; Yount, B.L.; Baric, R.S. Evaluation of a Recombination-Resistant Coronavirus as a Broadly Applicable, Rapidly Implementable Vaccine Platform. Commun. Biol. 2018, 1, 179. [Google Scholar] [CrossRef] [Green Version]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant Canine Coronaviruses Related to Transmissible Gastroenteritis Virus of Swine Are Circulating in Dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef] [Green Version]
- Tian, P.-F.; Jin, Y.-L.; Xing, G.; Qv, L.-L.; Huang, Y.-W.; Zhou, J.-Y. Evidence of Recombinant Strains of Porcine Epidemic Diarrhea Virus, United States, 2013. Emerg. Infect. Dis. 2014, 20, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Dudas, G.; Rambaut, A. MERS-CoV Recombination: Implications about the Reservoir and Potential for Adaptation. Virus Evol. 2016, 2, vev023. [Google Scholar] [CrossRef] [Green Version]
- Bobay, L.-M.; O’Donnell, A.C.; Ochman, H. Recombination Events Are Concentrated in the Spike Protein Region of Betacoronaviruses. PLoS Genet. 2020, 16, e1009272. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2021, 38, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- So, R.T.Y.; Chu, D.K.W.; Miguel, E.; Perera, R.A.P.M.; Oladipo, J.O.; Fassi-Fihri, O.; Aylet, G.; Ko, R.L.W.; Zhou, Z.; Cheng, M.-S.; et al. Diversity of Dromedary Camel Coronavirus HKU23 in African Camels Revealed Multiple Recombination Events among Closely Related Betacoronaviruses of the Subgenus Embecovirus. J. Virol. 2019, 93, e01236-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terada, Y.; Matsui, N.; Noguchi, K.; Kuwata, R.; Shimoda, H.; Soma, T.; Mochizuki, M.; Maeda, K. Emergence of Pathogenic Coronaviruses in Cats by Homologous Recombination between Feline and Canine Coronaviruses. PLoS ONE 2014, 9, e106534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytras, S.; Hughes, J.; Martin, D.; de Klerk, A.; Lourens, R.; Kosakovsky Pond, S.L.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rubnitz, J.; Subramani, S. The Minimum Amount of Homology Required for Homologous Recombination in Mammalian Cells. Mol. Cell. Biol. 1984, 4, 2253–2258. [Google Scholar] [CrossRef] [Green Version]
- Banner, L.R.; Lai, M.M. Random Nature of Coronavirus RNA Recombination in the Absence of Selection Pressure. Virology 1991, 185, 441–445. [Google Scholar] [CrossRef]
- Pollett, S.; Conte, M.A.; Sanborn, M.; Jarman, R.G.; Lidl, G.M.; Modjarrad, K.; Maljkovic Berry, I. A Comparative Recombination Analysis of Human Coronaviruses and Implications for the SARS-CoV-2 Pandemic. Sci. Rep. 2021, 11, 17365. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Sironi, M. Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses 2020, 12, 1313. [Google Scholar] [CrossRef]
- Stephens, J.C. On the Frequency of Undetectable Recombination Events. Genetics 1986, 112, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and Transmission of Interlineage Recombinants in the SARS-CoV-2 Pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef] [PubMed]
- VanInsberghe, D.; Neish, A.S.; Lowen, A.C.; Koelle, K. Recombinant SARS-CoV-2 Genomes Are Currently Circulating at Low Levels. Virus Evol. 2021, 7, veab059. [Google Scholar] [CrossRef]
- Turkahia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-Scale Phylogenomics Reveals Elevated Recombination Rates in the SARS-CoV-2 Spike Region. bioRxiv 2021. [Google Scholar] [CrossRef]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid Detection of Inter-Clade Recombination in SARS-CoV-2 with Bolotie. Genetics 2021, 218, iyab074. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through Recombination and Strong Purifying Selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Paraskevis, D.; Kostaki, E.G.; Magiorkinis, G.; Panayiotakopoulos, G.; Sourvinos, G.; Tsiodras, S. Full-Genome Evolutionary Analysis of the Novel Corona Virus (2019-NCoV) Rejects the Hypothesis of Emergence as a Result of a Recent Recombination Event. Infect. Genet. Evol. 2020, 79, 104212. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; Filip, I.; Chowdhury, R.; Maranas, C.D.; Sorger, P.K.; AlQuraishi, M.; Rabadan, R. Recombination and Lineage-Specific Mutations Linked to the Emergence of SARS-CoV-2. Genome Med. 2021, 13, 124. [Google Scholar] [CrossRef]
- Robertson, D.L.; Hahn, B.H.; Sharp, P.M. Recombination in AIDS Viruses. J. Mol. Evol. 1995, 40, 249–259. [Google Scholar] [CrossRef]
- Boniotti, M.B.; Papetti, A.; Lavazza, A.; Alborali, G.; Sozzi, E.; Chiapponi, C.; Faccini, S.; Bonilauri, P.; Cordioli, P.; Marthaler, D. Porcine Epidemic Diarrhea Virus and Discovery of a Recombinant Swine Enteric Coronavirus, Italy. Emerg. Infect. Dis. 2016, 22, 83–87. [Google Scholar] [CrossRef]
- Banerjee, A.; Doxey, A.C.; Tremblay, B.J.-M.; Mansfield, M.J.; Subudhi, S.; Hirota, J.A.; Miller, M.S.; McArthur, A.G.; Mubareka, S.; Mossman, K. Predicting the Recombination Potential of Severe Acute Respiratory Syndrome Coronavirus 2 and Middle East Respiratory Syndrome Coronavirus. J. Gen. Virol. 2020, 101, 1251–1260. [Google Scholar] [CrossRef]
- Yount, B.; Roberts, R.S.; Lindesmith, L.; Baric, R.S. Rewiring the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Transcription Circuit: Engineering a Recombination-Resistant Genome. Proc. Natl. Acad. Sci. USA 2006, 103, 12546–12551. [Google Scholar] [CrossRef] [Green Version]
- Burns, C.C.; Shaw, J.; Jorba, J.; Bukbuk, D.; Adu, F.; Gumede, N.; Pate, M.A.; Abanida, E.A.; Gasasira, A.; Iber, J.; et al. Multiple Independent Emergences of Type 2 Vaccine-Derived Polioviruses during a Large Outbreak in Northern Nigeria. J. Virol. 2013, 87, 4907–4922. [Google Scholar] [CrossRef] [Green Version]
- Guillot, S.; Caro, V.; Cuervo, N.; Korotkova, E.; Combiescu, M.; Persu, A.; Aubert-Combiescu, A.; Delpeyroux, F.; Crainic, R. Natural Genetic Exchanges between Vaccine and Wild Poliovirus Strains in Humans. J. Virol. 2000, 74, 8434–8443. [Google Scholar] [CrossRef] [Green Version]
- Pliaka, V.; Kyriakopoulou, Z.; Markoulatos, P. Risks Associated with the Use of Live-Attenuated Vaccine Poliovirus Strains and the Strategies for Control and Eradication of Paralytic Poliomyelitis. Expert Rev. Vaccines 2012, 11, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Karakasiliotis, I.; Paximadi, E.; Markoulatos, P. Evolution of a Rare Vaccine-Derived Multirecombinant Poliovirus. J. Gen. Virol. 2005, 86, 3137–3142. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulou, Z.; Pliaka, V.; Amoutzias, G.D.; Markoulatos, P. Recombination among Human Non-Polio Enteroviruses: Implications for Epidemiology and Evolution. Virus Genes 2015, 50, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-Scale Genomic Analysis Reveals Recurrent Patterns of Intertypic Recombination in Human Enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Schelle, B.; Karl, N.; Ludewig, B.; Siddell, S.G.; Thiel, V. Selective Replication of Coronavirus Genomes That Express Nucleocapsid Protein. J. Virol. 2005, 79, 6620–6630. [Google Scholar] [CrossRef] [Green Version]
- Sungsuwan, S.; Jongkaewwattana, A.; Jaru-Ampornpan, P. Nucleocapsid Proteins from Other Swine Enteric Coronaviruses Differentially Modulate PEDV Replication. Virology 2020, 540, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-Y.; Tsai, T.-L.; Lin, C.-N.; Lin, C.-H.; Wu, H.-Y. Interaction of Coronavirus Nucleocapsid Protein with the 5′- and 3′-Ends of the Coronavirus Genome Is Involved in Genome Circularization and Negative-Strand RNA Synthesis. FEBS J. 2019, 286, 3222–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e5. [Google Scholar] [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuñiga, S.; Enjuanes, L. RNA-RNA and RNA-Protein Interactions in Coronavirus Replication and Transcription. RNA Biol. 2011, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Züst, R.; Miller, T.B.; Goebel, S.J.; Thiel, V.; Masters, P.S. Genetic Interactions between an Essential 3′ Cis-Acting RNA Pseudoknot, Replicase Gene Products, and the Extreme 3′ End of the Mouse Coronavirus Genome. J. Virol. 2008, 82, 1214–1228. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Tsang, A.K.L.; Hui, S.-W.; Fan, R.Y.Y.; Martelli, P.; Yuen, K.-Y. Discovery of a Novel Bottlenose Dolphin Coronavirus Reveals a Distinct Species of Marine Mammal Coronavirus in Gammacoronavirus. J. Virol. 2014, 88, 1318–1331. [Google Scholar] [CrossRef] [Green Version]
- Elhaik, E.; Sabath, N.; Graur, D. The “Inverse Relationship between Evolutionary Rate and Age of Mammalian Genes” Is an Artifact of Increased Genetic Distance with Rate of Evolution and Time of Divergence. Mol. Biol. Evol. 2006, 23, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.F.; Bornberg-Bauer, E. Fact or Fiction: Updates on How Protein-Coding Genes Might Emerge de Novo from Previously Non-Coding DNA. F1000Research 2017, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Ouzounis, C.A. A Recent Origin of Orf3a from M Protein across the Coronavirus Lineage Arising by Sharp Divergence. Comput. Struct. Biotechnol. J. 2020, 18, 4093–4102. [Google Scholar] [CrossRef]
- Neches, R.Y.; Kyrpides, N.C.; Ouzounis, C.A. Atypical Divergence of SARS-CoV-2 Orf8 from Orf7a within the Coronavirus Lineage Suggests Potential Stealthy Viral Strategies in Immune Evasion. mBio 2021, 12, e03014-20. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; den Boon, J.A.; Horzinek, M.C.; Spaan, W.J. Comparison of the Genome Organization of Toro- and Coronaviruses: Evidence for Two Nonhomologous RNA Recombination Events during Berne Virus Evolution. Virology 1991, 180, 448–452. [Google Scholar] [CrossRef]
- Zeng, Q.; Langereis, M.A.; van Vliet, A.L.W.; Huizinga, E.G.; de Groot, R.J. Structure of Coronavirus Hemagglutinin-Esterase Offers Insight into Corona and Influenza Virus Evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 9065–9069. [Google Scholar] [CrossRef] [Green Version]
- Caprari, S.; Metzler, S.; Lengauer, T.; Kalinina, O.V. Sequence and Structure Analysis of Distantly-Related Viruses Reveals Extensive Gene Transfer between Viruses and Hosts and among Viruses. Viruses 2015, 7, 5388–5409. [Google Scholar] [CrossRef]
- Lang, Y.; Li, W.; Li, Z.; Koerhuis, D.; van den Burg, A.C.S.; Rozemuller, E.; Bosch, B.-J.; van Kuppeveld, F.J.M.; Boons, G.-J.; Huizinga, E.G.; et al. Coronavirus Hemagglutinin-Esterase and Spike Proteins Coevolve for Functional Balance and Optimal Virion Avidity. Proc. Natl. Acad. Sci. USA 2020, 117, 25759–25770. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.-D.; Zhang, H.-L.; Wang, M.-R.; Guan, X.-Q.; Holmes, E.C.; Zhang, Y.-Z. Extensive Genetic Diversity and Host Range of Rodent-Borne Coronaviruses. Virus Evol. 2020, 6, veaa078. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a Novel Coronavirus from a Beluga Whale by Using a Panviral Microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.-M.; Yang, Y.-L.; Xu, L.-D.; Wang, B.; Qin, P.; Huang, Y.-W. Porcine Torovirus (PToV)-A Brief Review of Etiology, Diagnostic Assays and Current Epidemiology. Front. Vet. Sci. 2019, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Shang, P.; Misra, S.; Hause, B.; Fang, Y. A Naturally Occurring Recombinant Enterovirus Expresses a Torovirus Deubiquitinase. J. Virol. 2017, 91, e00450-17. [Google Scholar] [CrossRef] [Green Version]
- Higdon, M.M.; Wahl, B.; Jones, C.B.; Rosen, J.G.; Truelove, S.A.; Baidya, A.; Nande, A.A.; ShamaeiZadeh, P.A.; Walter, K.K.; Feikin, D.R.; et al. A Systematic Review of COVID-19 Vaccine Efficacy and Effectiveness against SARS-CoV-2 Infection and Disease. medRxiv 2021. [Google Scholar] [CrossRef]
- Zimmer, C.; Corum, J.; Wee, S.-L. Coronavirus Vaccine Tracker. The New York Times. 2022. Available online: https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html (accessed on 23 November 2021).
- Ghasemiyeh, P.; Mohammadi-Samani, S.; Firouzabadi, N.; Dehshahri, A.; Vazin, A. A Focused Review on Technologies, Mechanisms, Safety, and Efficacy of Available COVID-19 Vaccines. Int. Immunopharmacol. 2021, 100, 108162. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. The Current Status of COVID-19 Vaccines. Front. Genome Ed. 2020, 2, 579297. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of Escape of SARS-CoV-2 Variant B.1.351 from Natural and Vaccine-Induced Sera. Cell 2021, 184, 2348–2361.e6. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody Evasion by the P.1 Strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 NCoV-19 COVID-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021, 384, 1885–1898. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased Risk of SARS-CoV-2 Reinfection Associated with Emergence of the Omicron Variant in South Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Martinez, D.R.; Schäfer, A.; Leist, S.R.; De la Cruz, G.; West, A.; Atochina-Vasserman, E.N.; Lindesmith, L.C.; Pardi, N.; Parks, R.; Barr, M.; et al. Chimeric Spike MRNA Vaccines Protect against Sarbecovirus Challenge in Mice. Science 2021, 373, 991–998. [Google Scholar] [CrossRef]
- Saunders, K.O.; Lee, E.; Parks, R.; Martinez, D.R.; Li, D.; Chen, H.; Edwards, R.J.; Gobeil, S.; Barr, M.; Mansouri, K.; et al. Neutralizing Antibody Vaccine for Pandemic and Pre-Emergent Coronaviruses. Nature 2021, 594, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-W.; Chia, W.-N.; Young, B.E.; Zhu, F.; Lim, B.-L.; Sia, W.-R.; Thein, T.-L.; Chen, M.I.-C.; Leo, Y.-S.; Lye, D.C.; et al. Pan-Sarbecovirus Neutralizing Antibodies in BNT162b2-Immunized SARS-CoV-1 Survivors. N. Engl. J. Med. 2021, 385, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-Reactive CD4+ T Cells Enhance SARS-CoV-2 Immune Responses upon Infection and Vaccination. Science 2021, 374, eabh1823. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.; Sheahan, T.; Heise, M.; Yount, B.; Davis, N.; Sims, A.; Suthar, M.; Harkema, J.; Whitmore, A.; Pickles, R.; et al. Vaccine Efficacy in Senescent Mice Challenged with Recombinant SARS-CoV Bearing Epidemic and Zoonotic Spike Variants. PLoS Med. 2006, 3, e525. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042.e21. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS-CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, e00647-20. [Google Scholar] [CrossRef]
- Harris, P.E.; Brasel, T.; Massey, C.; Herst, C.V.; Burkholz, S.; Lloyd, P.; Blankenberg, T.; Bey, T.M.; Carback, R.; Hodge, T.; et al. A Synthetic Peptide CTL Vaccine Targeting Nucleocapsid Confers Protection from SARS-CoV-2 Challenge in Rhesus Macaques. Vaccines 2021, 9, 520. [Google Scholar] [CrossRef]
- Dangi, T.; Class, J.; Palacio, N.; Richner, J.M.; Penaloza MacMaster, P. Combining Spike- and Nucleocapsid-Based Vaccines Improves Distal Control of SARS-CoV-2. Cell Rep. 2021, 36, 109664. [Google Scholar] [CrossRef]
- Hong, S.-H.; Oh, H.; Park, Y.W.; Kwak, H.W.; Oh, E.Y.; Park, H.-J.; Kang, K.W.; Kim, G.; Koo, B.-S.; Hwang, E.-H.; et al. Immunization with RBD-P2 and N Protects against SARS-CoV-2 in Nonhuman Primates. Sci. Adv. 2021, 7, eabg7156. [Google Scholar] [CrossRef]
- Matchett, W.E.; Joag, V.; Stolley, J.M.; Shepherd, F.K.; Quarnstrom, C.F.; Mickelson, C.K.; Wijeyesinghe, S.; Soerens, A.G.; Becker, S.; Thiede, J.M.; et al. Nucleocapsid Vaccine Elicits Spike-Independent SARS-CoV-2 Protective Immunity. J. Immunol. 2021, 207, 376–379. [Google Scholar] [CrossRef]
- Chiuppesi, F.; Nguyen, V.H.; Park, Y.; Contreras, H.; Karpinski, V.; Faircloth, K.; Nguyen, J.; Kha, M.; Johnson, D.; Martinez, J.; et al. Synthetic Multiantigen MVA Vaccine COH04S1 Protects Against SARS-CoV-2 in Syrian Hamsters and Non-Human Primates. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rammensee, H.-G.; Gouttefangeas, C.; Heidu, S.; Klein, R.; Preuß, B.; Walz, J.S.; Nelde, A.; Haen, S.P.; Reth, M.; Yang, J.; et al. Designing a SARS-CoV-2 T-Cell-Inducing Vaccine for High-Risk Patient Groups. Vaccines 2021, 9, 428. [Google Scholar] [CrossRef]
- Swadling, L.; Diniz, M.O.; Schmidt, N.M.; Amin, O.E.; Chandran, A.; Shaw, E.; Pade, C.; Gibbons, J.M.; Le Bert, N.; Tan, A.T.; et al. Pre-Existing Polymerase-Specific T Cells Expand in Abortive Seronegative SARS-CoV-2. Nature 2021. [Google Scholar] [CrossRef]
- Zhou, Y.-W.; Xie, Y.; Tang, L.-S.; Pu, D.; Zhu, Y.-J.; Liu, J.-Y.; Ma, X.-L. Therapeutic Targets and Interventional Strategies in COVID-19: Mechanisms and Clinical Studies. Signal. Transduct. Target. Ther. 2021, 6, 317. [Google Scholar] [CrossRef]
- MHRA First Oral Antiviral for COVID-19, Lagevrio (Molnupiravir), Approved by MHRA. Available online: https://www.gov.uk/government/news/first-oral-antiviral-for-covid-19-lagevrio-molnupiravir-approved-by-mhra (accessed on 5 November 2021).
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In Vitro Selection of Remdesivir Resistance Suggests Evolutionary Predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef] [PubMed]
- Molla, A.; Korneyeva, M.; Gao, Q.; Vasavanonda, S.; Schipper, P.J.; Mo, H.M.; Markowitz, M.; Chernyavskiy, T.; Niu, P.; Lyons, N.; et al. Ordered Accumulation of Mutations in HIV Protease Confers Resistance to Ritonavir. Nat. Med. 1996, 2, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Michelen, M.; Manoharan, L.; Elkheir, N.; Cheng, V.; Dagens, A.; Hastie, C.; O’Hara, M.; Suett, J.; Dahmash, D.; Bugaeva, P.; et al. Characterising Long COVID: A Living Systematic Review. BMJ Glob. Health 2021, 6, e005427. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Bowe, B.; Xie, Y. Long COVID after Breakthrough COVID-19: The Post-Acute Sequelae of Breakthrough COVID-19. ResearchSquare 2021. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary Analysis of the Delta and Delta Plus Variants of the SARS-CoV-2 Viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lubinski, B.; Frazier, L.E.; Phan, M.V.T.; Bugembe, D.L.; Tang, T.; Daniel, S.; Cotten, M.; Jaimes, J.A.; Whittaker, G.R. Spike Protein Cleavage-Activation Mediated by the SARS-CoV-2 P681R Mutation: A Case-Study from Its First Appearance in Variant of Interest (VOI) A.23.1 Identified in Uganda. bioRxiv 2021. [Google Scholar] [CrossRef]
- Smaoui, M.R.; Yahyaoui, H. Unraveling the Stability Landscape of Mutations in the SARS-CoV-2 Receptor-Binding Domain. Sci. Rep. 2021, 11, 9166. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Winkler, M.S.; Lier, M.; Schulz, S.; Jäck, H.-M.; Cossmann, A.; et al. The Spike Protein of SARS-CoV-2 Variant A.30 Is Heavily Mutated and Evades Vaccine-Induced Antibodies with High Efficiency. Cell Mol. Immunol. 2021, 18, 2673–2675. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Feng, Y.; Chen, H.; Luk, H.K.H.; Yang, W.-H.; Li, K.S.M.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, S.; Bouchama, A.; Mohammad Alharbi, B.; Rashid, M.; Saleem Khatlani, T.; Gaber, N.S.; Malik, S.S. SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence. Pathogens 2020, 9, 677. [Google Scholar] [CrossRef]
- Mostaghimi, D.; Valdez, C.N.; Larson, H.T.; Kalinich, C.C.; Iwasaki, A. Prevention of Host-to-Host Transmission by SARS-CoV-2 Vaccines. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Rella, S.A.; Kulikova, Y.A.; Dermitzakis, E.T.; Kondrashov, F.A. Rates of SARS-CoV-2 Transmission and Vaccination Impact the Fate of Vaccine-Resistant Strains. Sci. Rep. 2021, 11, 15729. [Google Scholar] [CrossRef]
- Mallapaty, S.; Callaway, E.; Kozlov, M.; Ledford, H.; Pickrell, J.; Van Noorden, R. How COVID Vaccines Shaped 2021 in Eight Powerful Charts. Nature 2021, 600, 580–583. [Google Scholar] [CrossRef]
- Zhou, P.; Shi, Z.-L. SARS-CoV-2 Spillover Events. Science 2021, 371, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. https://doi.org/10.3390/v14010078
Amoutzias GD, Nikolaidis M, Tryfonopoulou E, Chlichlia K, Markoulatos P, Oliver SG. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses. 2022; 14(1):78. https://doi.org/10.3390/v14010078
Chicago/Turabian StyleAmoutzias, Grigorios D., Marios Nikolaidis, Eleni Tryfonopoulou, Katerina Chlichlia, Panayotis Markoulatos, and Stephen G. Oliver. 2022. "The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2" Viruses 14, no. 1: 78. https://doi.org/10.3390/v14010078
APA StyleAmoutzias, G. D., Nikolaidis, M., Tryfonopoulou, E., Chlichlia, K., Markoulatos, P., & Oliver, S. G. (2022). The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses, 14(1), 78. https://doi.org/10.3390/v14010078