
In the Search of Marine Pestiviruses: First Case of Phocoena Pestivirus in a Belt Sea Harbour Porpoise

 , , , ,
, , , ,  , and
, and 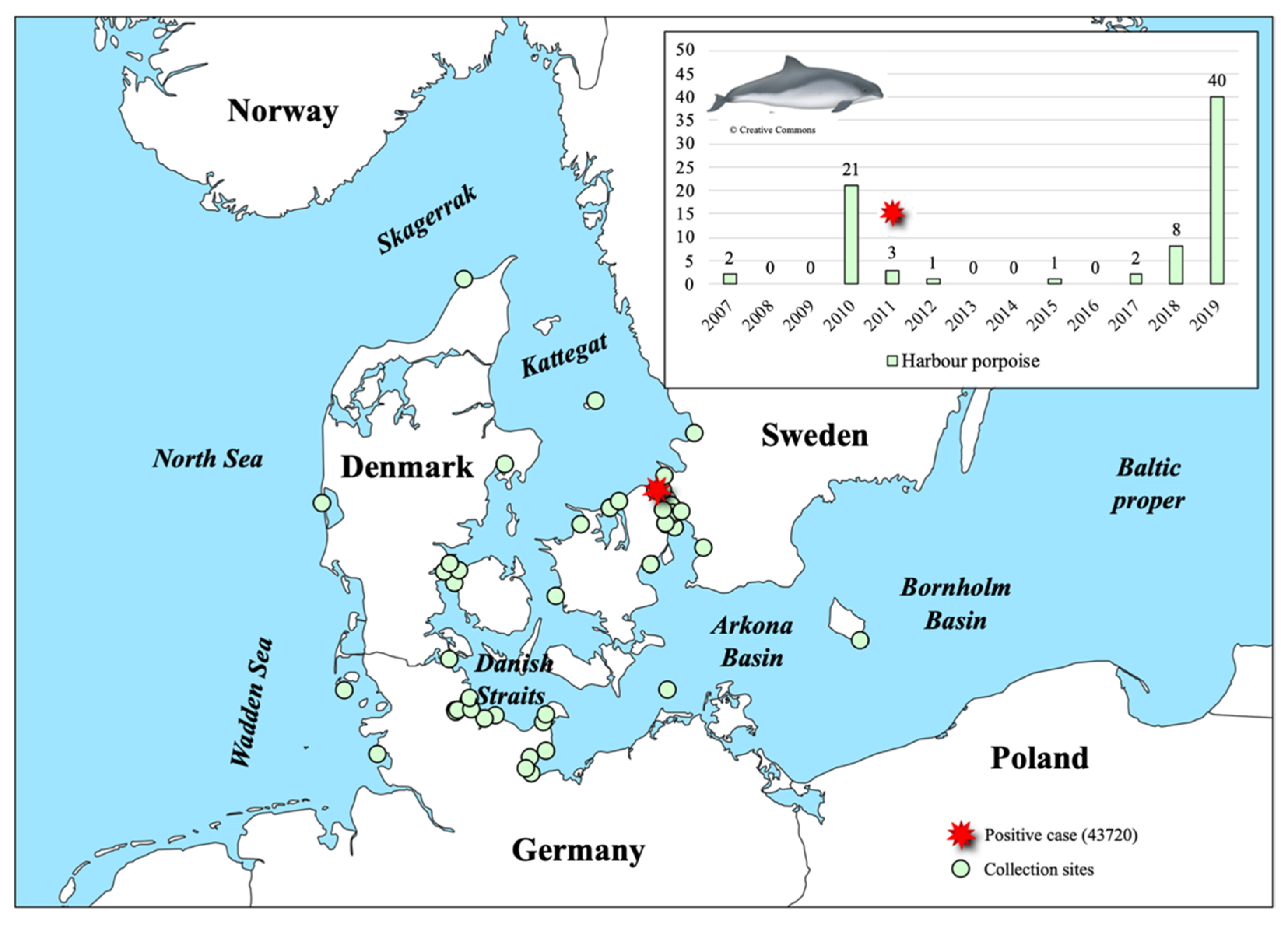{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Detection
2.2. Virus Extraction and Screening
2.3. Full Genome Sequencing and Phylogenetic Analyses
3. Results and Discussion
3.1. PhoPeV Pestivirus Detected in the Baltic Sea Region
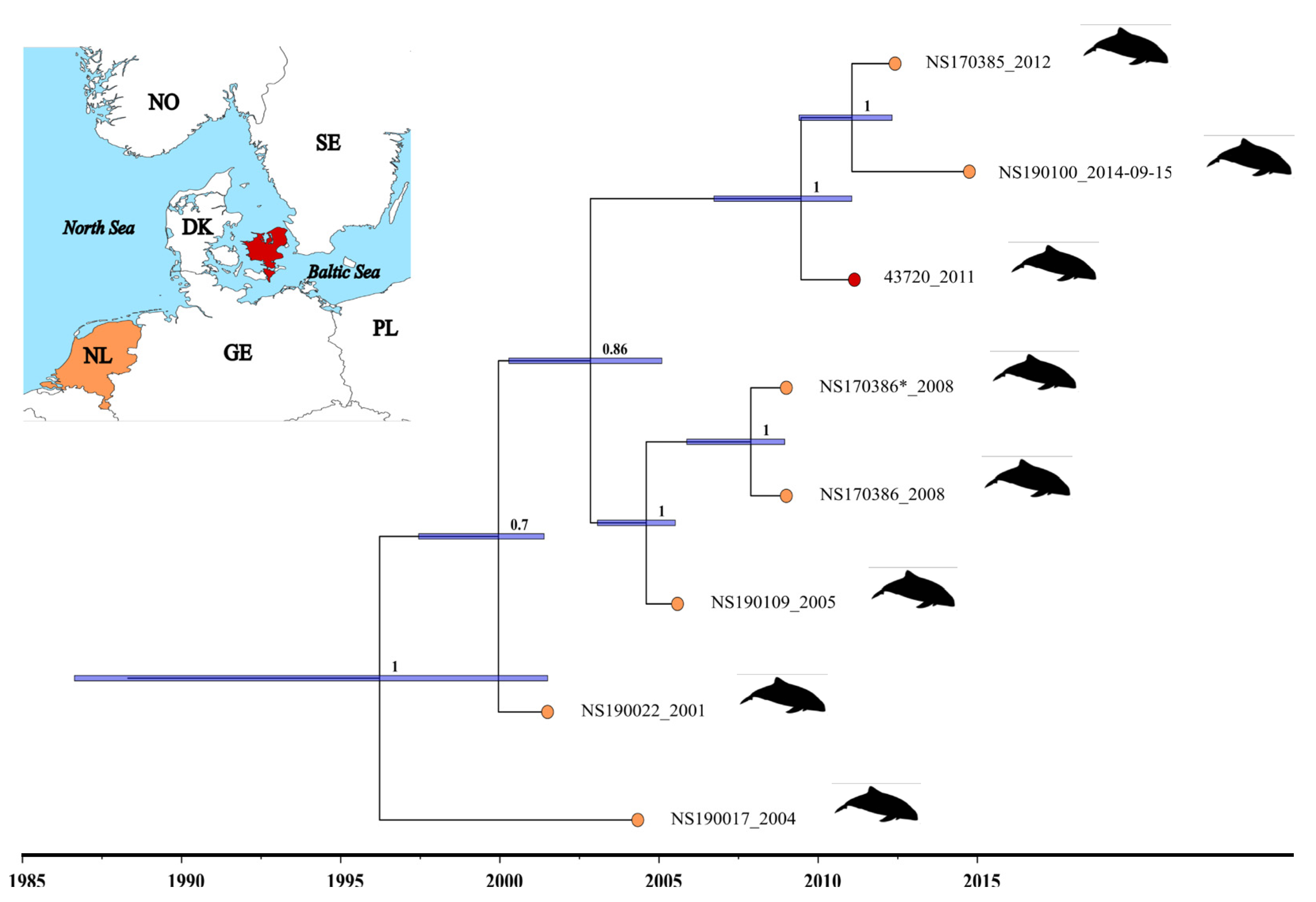
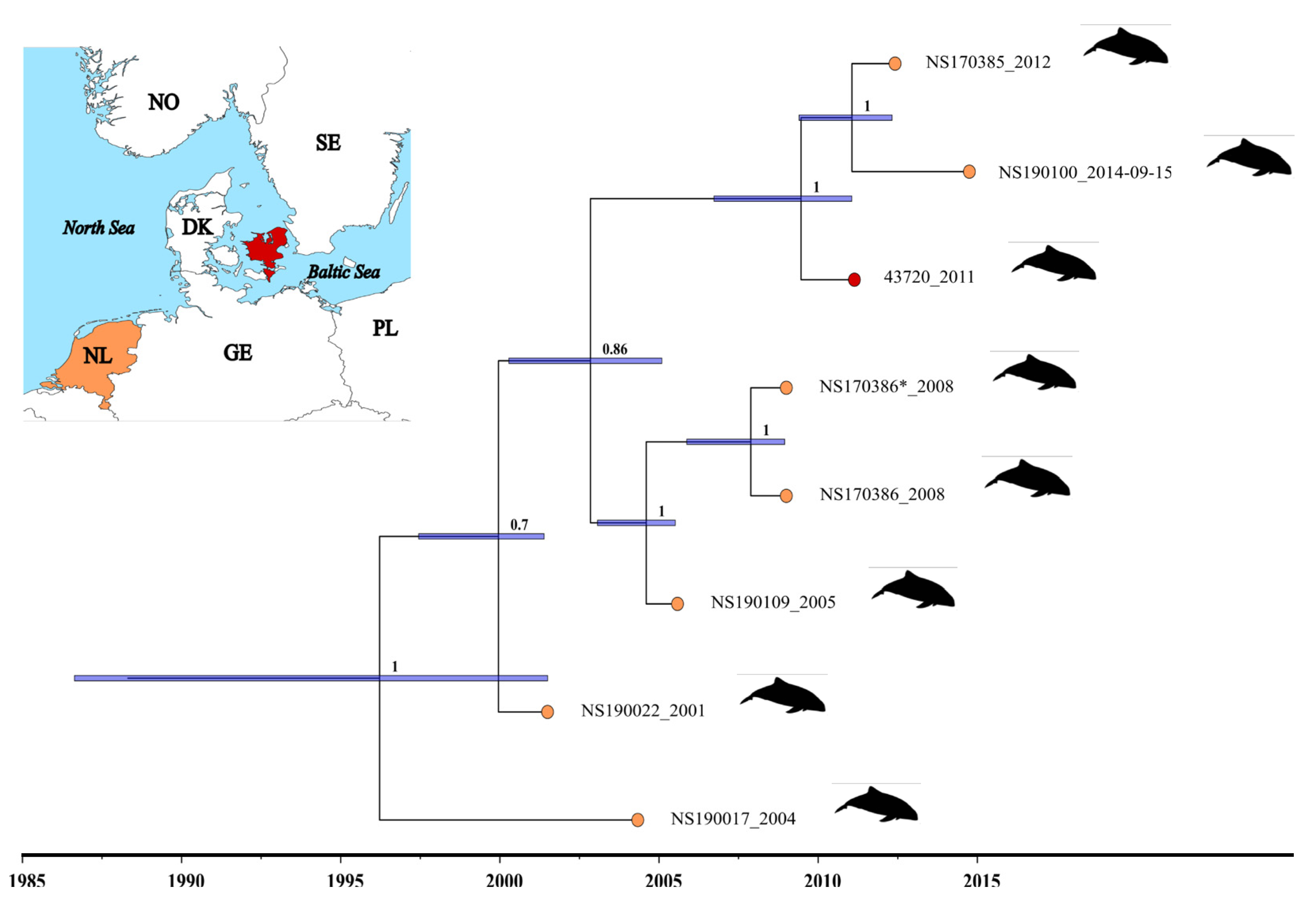
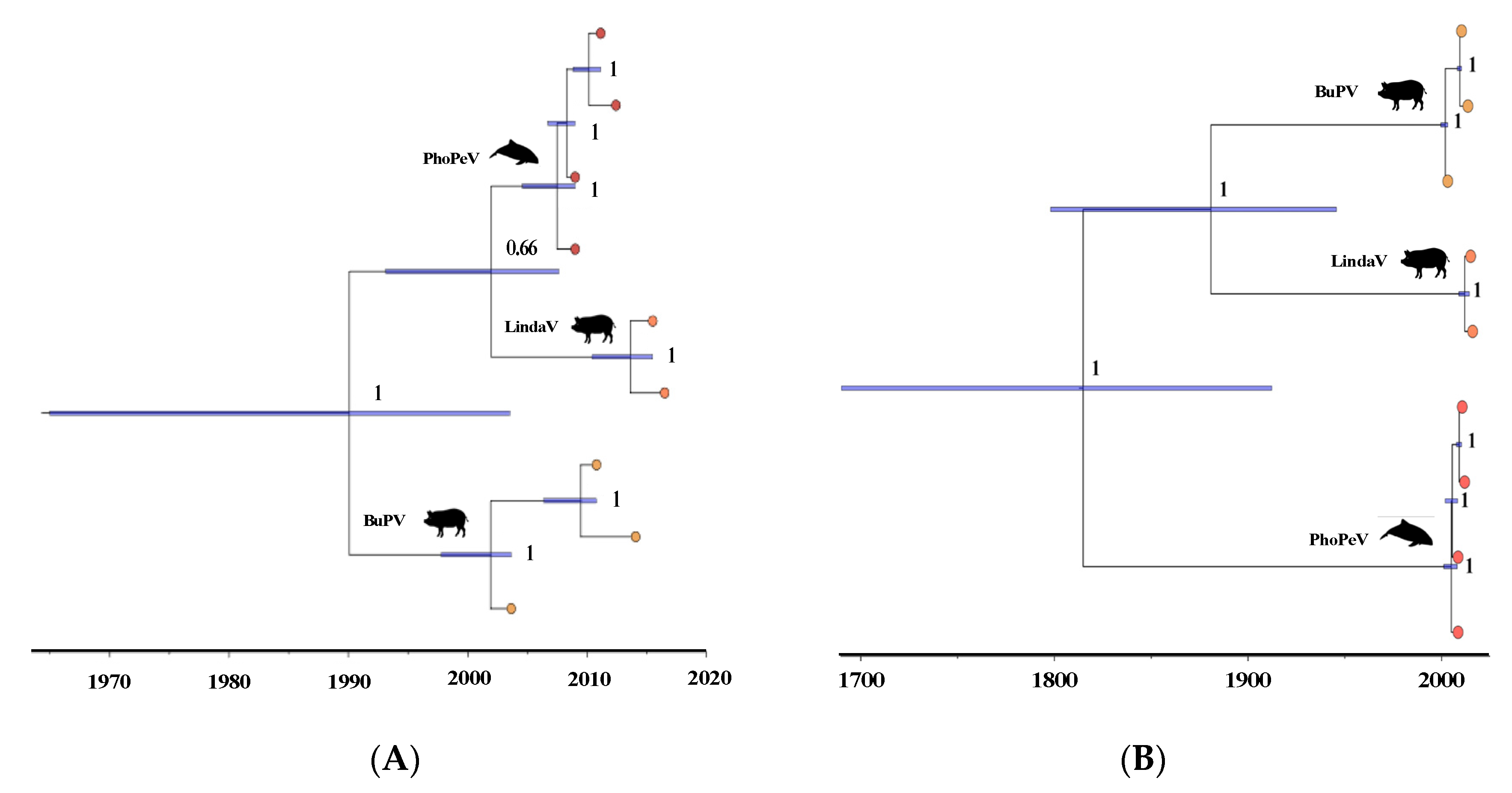
3.2. Determination of the Genome Sequence of PhoPeV Strain 43720 and the Phylogenetic Relationship among Porpoise Pestiviruses
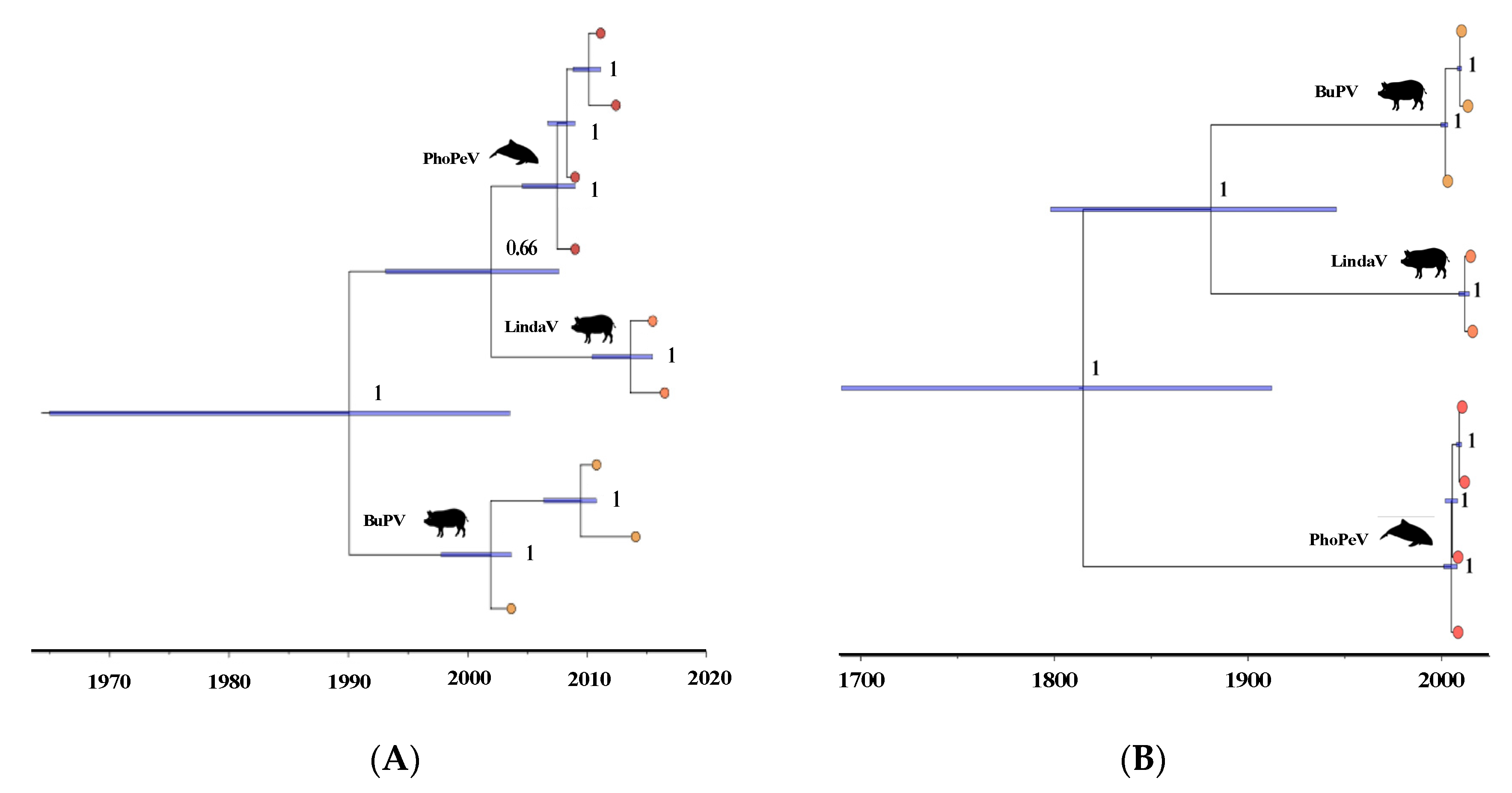
3.3. Phylogenetic Relationship of Terrestrial and Marine Pestiviruses
3.4. Pestiviruses in the Marine Ecosystem
3.5. Isolation of PhoPeV 43720 and Future Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Becher, P.; Moennig, V.; Tautz, N. Bovine Viral Diarrhea, Border Disease, and Classical Swine Fever Viruses; Elsevier Ltd.: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Smith, D.B.; Meyers, G.; Bukh, J.; Gould, E.A.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; Simmonds, P.; et al. Proposed Revision to the Taxonomy of the Genus Pestivirus, Family Flaviviridae. J. Gen. Virol. 2017, 98, 2106–2112. [Google Scholar] [CrossRef]
- Moennig, V.; Becher, P. Pestivirus Control Programs: How Far Have We Come and Where Are We Going? Anim. Heal. Res. Rev. 2015, 16, 83–87. [Google Scholar] [CrossRef]
- Moennig, V.; Becher, P. Control of Bovine Viral Diarrhea. Pathogens 2018, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Postel, A.; Smith, D.B.; Becher, P. Proposed Update to the Taxonomy of Pestiviruses: Eight Additional Species within the Genus Pestivirus, Family Flaviviridae. Viruses 2021, 13, 1542. [Google Scholar] [CrossRef]
- Lamp, B.; Schwarz, L.; Högler, S.; Riedel, C.; Sinn, L.; Rebel-Bauder, B.; Weissenböck, H.; Ladinig, A.; Rümenapf, T. Novel Pestivirus Species in Pigs, Austria, 2015. Emerg. Infect. Dis. 2017, 23, 1176–1179. [Google Scholar] [CrossRef]
- Thabti, F.; Letellier, C.; Hammami, S.; Pépin, M.; Ribière, M.; Mesplède, A.; Kerkhofs, P.; Russo, P. Detection of a Novel Border Disease Virus Subgroup in Tunisian Sheep. Arch. Virol. 2005, 150, 215–229. [Google Scholar] [CrossRef]
- Postel, A.; Schmeiser, S.; Oguzoglu, T.C.; Indenbirken, D.; Alawi, M.; Fischer, N.; Grundhoff, A.; Becher, P. Close Relationship of Ruminant Pestiviruses and Classical Swine Fever Virus. Emerg. Infect. Dis. 2015, 21, 668–672. [Google Scholar] [CrossRef]
- Ciulli, S.; Purpari, G.; Agnello, S.; Di Marco, P.; Di Bella, S.; Volpe, E.; Mira, F.; de Aguiar Saldanha Pinheiro, A.C.; Vullo, S.; Guercio, A. Evidence for Tunisian-Like Pestiviruses Presence in Small Ruminants in Italy Since 2007. Transbound. Emerg. Dis. 2017, 64, 1243–1253. [Google Scholar] [CrossRef]
- Sozzi, E.; Lavazza, A.; Gaffuri, A.; Bencetti, F.C.; Prosperi, A.; Lelli, D.; Chiapponi, C.; Moreno, A. Isolation and Full-Length Sequence Analysis of a Pestivirus from Aborted Lamb Fetuses in Italy. Viruses 2019, 11, 744. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.; Postel, A.; Wiedemann, A.; Cagatay, G.N.; Ciulli, S.; Guercio, A.; Becher, P. Comparative Analysis of Tunisian Sheep-like Virus, Bungowannah Virus and Border Disease Virus Infection in the Porcine Host. Viruses 2021, 13, 1539. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liu, B.; Du, J.; Zhang, J.; Lu, L.; Zhu, G.; Han, Y.; Su, H.; Yang, L.; Zhang, S.; et al. Discovery of Diverse Rodent and Bat Pestiviruses with Distinct Genomic and Phylogenetic Characteristics in Several Chinese Provinces. Front. Microbiol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of Zoonotic Pathogens and Characterization of Novel Viruses Carried by Commensal Rattus Norvegicus in New York City. mBio 2014, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.H.; Lin, X.D.; Chen, Y.M.; Xie, C.G.; Tan, Z.Z.; Zhou, J.J.; Chen, S.; Holmes, E.C.; Zhang, Y.Z. Newly Identified Viral Genomes in Pangolins with Fatal Disease. Virus Evol. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Jo, W.K.; van Elk, C.; van de Bildt, M.; van Run, P.; Petry, M.; Jesse, S.T.; Jung, K.; Ludlow, M.; Kuiken, T.; Osterhaus, A. An Evolutionary Divergent Pestivirus Lacking the Npro Gene Systemically Infects a Whale Species. Emerg. Microbes Infect. 2019, 8, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Tautz, N.; Tews, B.A.; Meyers, G. The Molecular Biology of Pestiviruses, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 93. [Google Scholar] [CrossRef]
- De Groof, A.; Deijs, M.; Guelen, L.; Van Grinsven, L.; Van Os-Galdos, L.; Vogels, W.; Derks, C.; Cruijsen, T.; Geurts, V.; Vrijenhoek, M.; et al. Atypical Porcine Pestivirus: A Possible Cause of Congenital Tremor Type A-II in Newborn Piglets. Viruses 2016, 8, 271. [Google Scholar] [CrossRef]
- Postel, A.; Hansmann, F.; Baechlein, C.; Fischer, N.; Alawi, M.; Grundhoff, A.; Derking, S.; Tenhündfeld, J.; Pfankuche, V.M.; Herder, V.; et al. Presence of Atypical Porcine Pestivirus (APPV) Genomes in Newborn Piglets Correlates with Congenital Tremor. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Finlaison, D.S.; Kirkland, P.D. The Outcome of Porcine Foetal Infection with Bungowannah Virus Is Dependent on the Stage of Gestation at Which Infection Occurs. Part 2: Clinical Signs and Gross Pathology. Viruses 2020, 12, 873. [Google Scholar] [CrossRef]
- Sveegaard, S.; Galatius, A.; Dietz, R.; Kyhn, L.; Koblitz, J.C.; Amundin, M.; Nabe-Nielsen, J.; Sinding, M.H.S.; Andersen, L.W.; Teilmann, J. Defining Management Units for Cetaceans by Combining Genetics, Morphology, Acoustics and Satellite Tracking. Glob. Ecol. Conserv. 2015, 3, 839–850. [Google Scholar] [CrossRef] [Green Version]
- ASCOBANS. Draft Recovery Plan for Baltic Harbour Porpoises (Jastarnia Plan); ASCOBANS: Bonn, Germany, 2002. [Google Scholar]
- Benke, H.; Bräger, S.; Dähne, M.; Gallus, A.; Hansen, S.; Honnef, C.G.; Jabbusch, M.; Koblitz, J.C.; Krügel, K.; Liebschner, A.; et al. Baltic Sea Harbour Porpoise Populations: Status and Conservation Needs Derived from Recent Survey Results. Mar. Ecol. Prog. Ser. 2014, 495, 275–290. [Google Scholar] [CrossRef] [Green Version]
- Fischer, N.; Indenbirken, D.; Meyer, T.; Lütgehetmann, M.; Lellek, H.; Spohn, M.; Aepfelbacher, M.; Alawi, M.; Grundhoff, A. Evaluation of Unbiased Next-Generation Sequencing of RNA (RNA-Seq) as a Diagnostic Method in Influenza Virus-Positive Respiratory Samples. J. Clin. Microbiol. 2015, 53, 2238–2250. [Google Scholar] [CrossRef] [Green Version]
- Alawi, M.; Burkhardt, L.; Indenbirken, D.; Reumann, K.; Christopeit, M.; Kröger, N.; Lütgehetmann, M.; Aepfelbacher, M.; Fischer, N.; Grundhoff, A. DAMIAN: An Open Source Bioinformatics Tool for Fast, Systematic and Cohort Based Analysis of Microorganisms in Diagnostic Samples. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: A Suite of Phylogenetic Analysis Tools for Evolutionary Biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Zhang, J.; Pei, N.; Mi, X. Phylotools: Phylogenetic Tools for Eco-Phylogenetics. v. 0.2.2. 2017. Available online: https://cran.r-project.org/package=phylotools.
- Knudsen, S.W.; Choat, J.H.; Clements, K.D. The Herbivorous Fish Family Kyphosidae (Teleostei: Perciformes) Represents a Recent Radiation from Higher Latitudes. J. Biogeogr. 2019, 46, 2067–2080. [Google Scholar] [CrossRef]
- Kück, P.; Meusemann, K. FASconCAT; Zoologisches Forschungsmuseum: Bonn, Germany, 2010. [Google Scholar]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Surchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer.
- Goldman, N.; Yang, Z. A Codon-Based Model of Nucleotide Substitution for Protein-Coding DNA Sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Düx, A.; Lequime, S.; Patrono, L.V.; Vrancken, B.; Boral, S.; Gogarten, J.F.; Hilbig, A.; Horst, D.; Merkel, K.; Prepoint, B.; et al. Measles Virus and Rinderpest Virus Divergence Dated to the Sixth Century BCE. Science 2020, 368, 1367–1370. [Google Scholar] [CrossRef]
- Mühlemann, B.; Vinner, L.; Margaryan, A.; Wilhelmson, H.; de la Fuente Castro, C.; Allentoft, M.E.; de Barros Damgaard, P.; Hansen, A.J.; Holtsmark Nielsen, S.; Strand, L.M.; et al. Diverse Variola Virus (Smallpox) Strains Were Widespread in Northern Europe in the Viking Age. Science 2020, 369, 6502. [Google Scholar] [CrossRef]
- McOrist, S.; Thornton, E.; Peake, A.; Walker, R.; Robson, S.; Finlaison, D.; Kirkland, P.; Reece, R.; Ross, A.; Walker, K.; et al. An Infectious Myocarditis Syndrome Affecting Late-Term and Neonatal Piglets. Aust. Vet. J. 2004, 82, 509–511. [Google Scholar]
- Kirkland, P.D.; Read, A.J.; Frost, M.J.; Finlaison, D.S. Bungowannah Virus—A Probable New Species of Pestivirus—What Have We Found in the Last 10 Years? Anim. Heal. Res. Rev. 2015, 16, 60–63. [Google Scholar] [CrossRef]
- Cagatay, G.N.; Antos, A.; Meyer, D.; Maistrelli, C.; Keuling, O.; Becher, P.; Postel, A. Frequent Infection of Wild Boar with Atypical Porcine Pestivirus (APPV). Transbound. Emerg. Dis. 2018, 65, 1087–1093. [Google Scholar] [CrossRef]
- Michelitsch, A.; Dalmann, A.; Wernike, K.; Reimann, I.; Beer, M. Seroprevalences of Newly Discovered Porcine Pestiviruses in German Pig Farms. Vet. Sci. 2019, 6, 86. [Google Scholar] [CrossRef] [Green Version]
- Kiesler, A.; Plankensteiner, J.; Schwarz, L.; Riedel, C.; Seitz, K.; Mötz, M.; Ladinig, A.; Lamp, B.; Rümenapf, T. Prevalence of Linda Virus Neutralizing Antibodies in the Austrian Pig Population. Viruses 2021, 13, 1001. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokholm, I.; Fischer, N.; Baechlein, C.; Postel, A.; Galatius, A.; Kyhn, L.A.; Thøstesen, C.B.; Persson, S.; Siebert, U.; Olsen, M.T.; et al. In the Search of Marine Pestiviruses: First Case of Phocoena Pestivirus in a Belt Sea Harbour Porpoise. Viruses 2022, 14, 161. https://doi.org/10.3390/v14010161
Stokholm I, Fischer N, Baechlein C, Postel A, Galatius A, Kyhn LA, Thøstesen CB, Persson S, Siebert U, Olsen MT, et al. In the Search of Marine Pestiviruses: First Case of Phocoena Pestivirus in a Belt Sea Harbour Porpoise. Viruses. 2022; 14(1):161. https://doi.org/10.3390/v14010161
Chicago/Turabian StyleStokholm, Iben, Nicole Fischer, Christine Baechlein, Alexander Postel, Anders Galatius, Line Anker Kyhn, Charlotte Bie Thøstesen, Sara Persson, Ursula Siebert, Morten Tange Olsen, and et al. 2022. "In the Search of Marine Pestiviruses: First Case of Phocoena Pestivirus in a Belt Sea Harbour Porpoise" Viruses 14, no. 1: 161. https://doi.org/10.3390/v14010161
APA StyleStokholm, I., Fischer, N., Baechlein, C., Postel, A., Galatius, A., Kyhn, L. A., Thøstesen, C. B., Persson, S., Siebert, U., Olsen, M. T., & Becher, P. (2022). In the Search of Marine Pestiviruses: First Case of Phocoena Pestivirus in a Belt Sea Harbour Porpoise. Viruses, 14(1), 161. https://doi.org/10.3390/v14010161