
Genomic Instability and DNA Damage Repair Pathways Induced by Human Papillomaviruses



Abstract
1. Introduction
2. Genome Organization
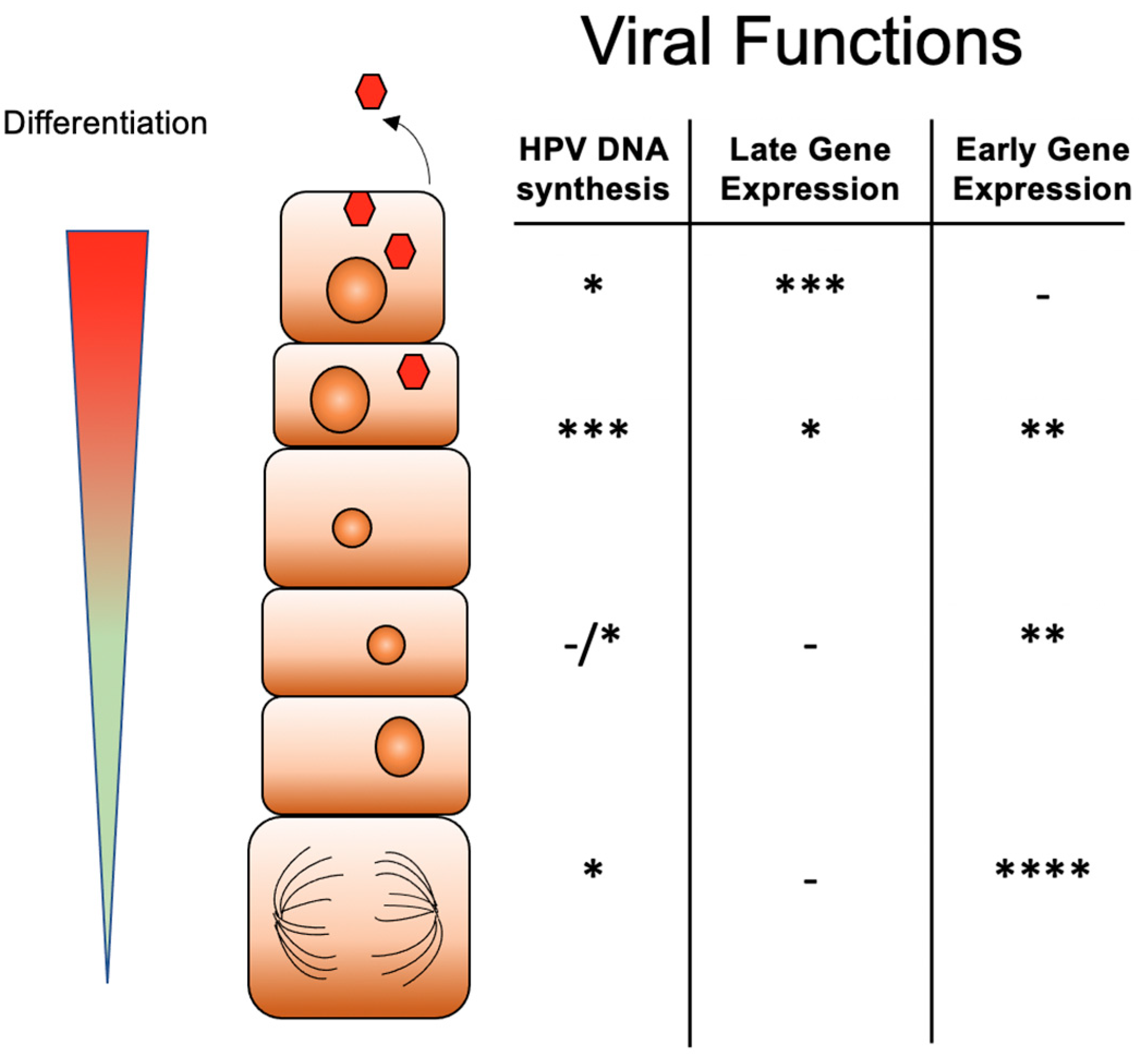
3. The Differentiation-Dependent Life Cycle of HPVs
4. Host DNA Damage Repair Pathways
5. The DNA Damage Response and the HPV Life Cycle
6. Progression to Cancer
7. Mutation Signatures in HPV Positive Cancers
8. APOBECs and HPV Positive Lesions
9. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pastrana, D.V.; Peretti, A.; Welch, N.L.; Borgogna, C.; Olivero, C.; Badolato, R.; Notarangelo, L.D.; Gariglio, M.; FitzGerald, P.C.; McIntosh, C.M.; et al. Metagenomic Discovery of 83 New Human Papillomavirus Types in Patients with Immunodeficiency. Msphere 2018, 3, e00645-18. [Google Scholar] [CrossRef] [PubMed]
- Stokley, S.; Jeyarajah, J.; Yankey, D.; Cano, M.; Gee, J.; Roark, J.; Curtis, R.; Markowitz, L. Human papillomavirus vaccination coverage among adolescents, 2007-2013, and postlicensure vaccine safety monitoring, 2006–2014—United States. MMWR Morb. Mortal. Wkly. Rep. 2014, 63, 620–624. [Google Scholar] [PubMed]
- Steenbergen, R.D.; Snijders, P.J.; Heideman, D.A.; Meijer, C.J. Clinical implications of (epi)genetic changes in HPV-induced cervical precancerous lesions. Nat. Rev. Cancer 2014, 14, 395–405. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Mork, J.; Lie, A.K.; Glattre, E.; Hallmans, G.; Jellum, E.; Koskela, P.; Moller, B.; Pukkala, J.; Schiller, J.; Youngman, L.; et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2001, 344, 1125–1131. [Google Scholar] [CrossRef]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Castellsague, X.; Alemany, L.; Quer, M.; Halec, G.; Quiros, B.; Tous, S.; Clavero, O.; Alos, L.; Biegner, T.; Szafarowski, T.; et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. J. Natl. Cancer Inst. 2016, 108, djv403. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Snijders, P.J.; Steenbergen, R.D.; Heideman, D.A.; Meijer, C.J. HPV-mediated cervical carcinogenesis: Concepts and clinical implications. J. Pathol. 2006, 208, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Ustav, M.; Stenlund, A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991, 10, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Mohr, I.J.; Clark, R.; Sun, S.; Androphy, E.J.; MacPherson, P.; Botchan, M.R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 1990, 250, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Martin, S.R.; Jackson, D.J.; Khan, J.; Isaacson, E.R.; Calder, L.; et al. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1--E4 protein and provides a molecular basis for its accumulation. J. Virol. 2008, 82, 8196–8203. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Griffin, H.; Southern, S.; Jackson, D.; Martin, A.; McIntosh, P.; Davy, C.; Masterson, P.J.; Walker, P.A.; Laskey, P.; et al. Functional analysis of the human papillomavirus type 16 E1=E4 protein provides a mechanism for in vivo and in vitro keratin filament reorganization. J. Virol. 2004, 78, 821–833. [Google Scholar] [CrossRef]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Laimins, L.A. Human papillomavirus life cycle: Active and latent phases. Semin. Cancer Biol. 1999, 9, 379–386. [Google Scholar] [CrossRef]
- Del Vecchio, A.M.; Romanczuk, H.; Howley, P.M.; Baker, C.C. Transient replication of human papillomavirus DNAs. J. Virol. 1992, 66, 5949–5958. [Google Scholar] [CrossRef]
- Rohlfs, M.; Winkenbach, S.; Meyer, S.; Rupp, T.; Durst, M. Viral transcription in human keratinocyte cell lines immortalized by human papillomavirus type-16. Virology 1991, 183, 331–342. [Google Scholar] [CrossRef]
- Egawa, N.; Nakahara, T.; Ohno, S.; Narisawa-Saito, M.; Yugawa, T.; Fujita, M.; et al. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 2012, 86, 3276–3283. [Google Scholar] [CrossRef]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D. The DNA damage response, immunity and cancer. Semin. Cancer Biol. 2006, 16, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.T.; Lane, W.; Kastan, M.; D’Andrea, A. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002, 109, 459–472. [Google Scholar] [CrossRef]
- Grompe, M. FANCD2: A branch-point in DNA damage response? Nat. Med. 2002, 8, 555–556. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 2018, 9, e00064-18. [Google Scholar] [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Gunasekharan, V.; Satsuka, A.; Laimins, L.A. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog. 2015, 11, e1004763. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene to Activate the ATR Pathway and Promote Human Papillomavirus Replication. mBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Li, Y.; Kaminski, P.J.; Andrade, J.; Laimins, L.A. Pathogenesis of Human Papillomaviruses Requires the ATR/p62 Autophagy-Related Pathway. mBio 2020, 11, e01628-20. [Google Scholar] [CrossRef]
- Spriggs, C.C.; Laimins, L.A. FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication. mBio 2017, 8, e02340-16. [Google Scholar] [CrossRef]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Kaminski, P.; Hong, S.; Kono, T.; Hoover, P.; Laimins, L. Topoisomerase 2beta Induces DNA Breaks To Regulate Human Papillomavirus Replication. mBio 2021, 12, e00005-21. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Blanco, L.Z.; Maniar, K.P.; Laimins, L.A. Expression of HPV-induced DNA Damage Repair Factors Correlates with CIN Progression. Int. J. Gynecol. Pathol. 2019, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rietbergen, M.M.; Brakenhoff, R.H.; Bloemena, E.; Witte, B.I.; Snijders, P.J.; Heideman, D.A.; Boon, D.; Koljenovic, S.; Battenberg, R.; Leemans, C. Human papillomavirus detection and comorbidity: Critical issues in selection of patients with oropharyngeal cancer for treatment De-escalation trials. Ann. Oncol. 2013, 24, 2740–2745. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.; Newcombe, R.G.; Green, A.C.; Kelly, C.; Noel Gill, O.; Hall, G.; Fiander, A.; Pirotte, E.; Hibbitts, S.; Homer, J. Human papillomavirus infection is rare in nonmalignant tonsil tissue in the UK: Implications for tonsil cancer precursor lesions. Int. J. Cancer 2014, 135, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Sehr, P.; Waterboer, T.; Leivo, I.; Pawlita, M.; Vaheri, A.; Aaltonen, L. Presence of DNA of human papillomavirus 16 but no other types in tumor-free tonsillar tissue. J. Clin. Microbiol. 2005, 43, 1408–1410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Combes, J.D.; Dalstein, V.; Gheit, T.; Clifford, G.M.; Tommasino, M.; Clavel, C.; Lacau, S.; Guily, J.; Franeschi, S.; SPLIT study group. Prevalence of human papillomavirus in tonsil brushings and gargles in cancer-free patients: The SPLIT study. Oral. Oncol. 2017, 66, 52–57. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Gwin, T.K.; McLaughlin-Drubin, M.E. p16 in HPV-associated cancers. Oncotarget 2013, 4, 1864–1865. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- Kim, S.H.; Koo, B.S.; Kang, S.; Park, K.; Kim, H.; Lee, K.R.; Lee, M.; Kim, J.; Choi, E.; Cho, N. HPV integration begins in the tonsillar crypt and leads to the alteration of p16, EGFR and c-myc during tumor formation. Int. J. Cancer 2007, 120, 1418–1425. [Google Scholar] [CrossRef]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agarwal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell. Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.N.; Van Waes, C.; Seiwert, T.Y. Genetic Landscape of Human Papillomavirus-Associated Head and Neck Cancer and Comparison to Tobacco-Related Tumors. J. Clin. Oncol. 2015, 33, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Goila-Gaur, R.; Strebel, K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 2008, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Wallace, N.A.; Munger, K. The curious case of APOBEC3 activation by cancer-associated human papillomaviruses. PLoS Pathog. 2018, 14, e1006717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.; Carey, T.; Chepeha, D.; Prince, M.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.; Howley, P.; Harris, R. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio 2014, 5, e02234-14. [Google Scholar] [CrossRef]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Yugawa, T.; Kiyono, T.; Nishina, H.; Kukimoto, I. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J. Virol. 2017, 91, e02413-16. [Google Scholar] [CrossRef]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem. Biophys. Res. Commun. 2015, 460, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; McCann, J.L.; Starrett, G.J.; Kosyakovsky, L.; Luengas, E.M.; Molan, A.M.; Burns, M.; McDougle, R.; Parker, P.; Brown, W.; et al. The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015, 75, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Mo, D.; Liu, H.; Veena, M.S.; Srivatsan, E.S.; Massoumi, R.; Rettig, M. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 2008, 14, 394–407. [Google Scholar] [CrossRef]
- Fischer, M.; Steiner, L.; Engeland, K. The transcription factor p53: Not a repressor, solely an activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Kranjec, C.; Farzan, R.; Leach, D.A.; et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res. 2017, 45, 11056–11069. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92, e01318-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Imayasu, M.; Shigenobu, S.; Nishiyama, T.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Wakae, K.; Wakisaka, N.; Nakanishi, Y.; Ishikawa, K.; Komori, T.; Nishiyama, T.; Yamguchi, K.; Shigenobu, S.; Miramatsu, M.; et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene 2017, 36, 1687–1697. [Google Scholar] [CrossRef]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.; Fargo, D.; Mieczkowski, P.; et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Hoover, P.; Poropatich, K.; Paunesku, T.; Mittal, B.B.; Samant, S.; Laimins, L. Activation of DNA damage repair factors in HPV positive oropharyngeal cancers. Virology 2020, 547, 27–34. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Roberts, S.A.; Malc, E.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Malkova, A. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014, 7, 1640–1648. [Google Scholar] [CrossRef]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Kimczak, L.; Kryukov, G.; Malc, E.; Mieczkowski, P.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef]
- Kanu, N.; Cerone, M.A.; Goh, G.; Zalmas, L.P.; Bartkova, J.; Dietzen, M.; McGranahan, N.; Rogers, R.; Law, E.; Gromova, I.; et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Nikkila, J.; Kumar, R.; Campbell, J.; Brandsma, I.; Pemberton, H.; Walberg, F.; Nagy, K.; Scheer, I.; Vetessy, B.; Serebrenik, A.; et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br. J. Cancer 2017, 117, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Budagyan, K.; Hayer, K.E.; Reed, M.A.; Savani, M.R.; Wertheim, G.B.; Weitzman, M. Cytosine Deaminase APOBEC3A Sensitizes Leukemia Cells to Inhibition of the DNA Replication Checkpoint. Cancer Res. 2017, 77, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Marafioti, T.; Henry, J.; Van Allen, E.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.; Vogel, R.; van Gelder, M.; Sweep, F.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [PubMed]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.; Somasundaran, M.; Yilmaz, N.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.; Starett, G.; Duffy, J.; Demir, O.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| DDR Factor | Binds HPV Genome Replication | Stable Replication | Required for Amplification |
|---|---|---|---|
| pCHK2 | Yes | No | Yes |
| pCHK1 | ND | Yes | Yes |
| RAD51 | Yes | ND | Yes |
| BRCA1 | Yes | ND | Yes |
| FANCD2 | Yes | Yes | Indirectly |
| γH2AX | Yes | ND | ND |
| pSMC1 | Yes | ND | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kono, T.; Laimins, L. Genomic Instability and DNA Damage Repair Pathways Induced by Human Papillomaviruses. Viruses 2021, 13, 1821. https://doi.org/10.3390/v13091821
Kono T, Laimins L. Genomic Instability and DNA Damage Repair Pathways Induced by Human Papillomaviruses. Viruses. 2021; 13(9):1821. https://doi.org/10.3390/v13091821
Chicago/Turabian StyleKono, Takeyuki, and Laimonis Laimins. 2021. "Genomic Instability and DNA Damage Repair Pathways Induced by Human Papillomaviruses" Viruses 13, no. 9: 1821. https://doi.org/10.3390/v13091821
APA StyleKono, T., & Laimins, L. (2021). Genomic Instability and DNA Damage Repair Pathways Induced by Human Papillomaviruses. Viruses, 13(9), 1821. https://doi.org/10.3390/v13091821