
The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Genomic DNA and Plasmid DNA Extraction
2.3. Polymerase Chain Reaction
2.4. Gel Electrophoresis
2.5. Site-Directed Mutagenesis (SDM) of Pf4r
2.6. Electrophoretic Mobility Shift Assay (EMSA) of Pf4r
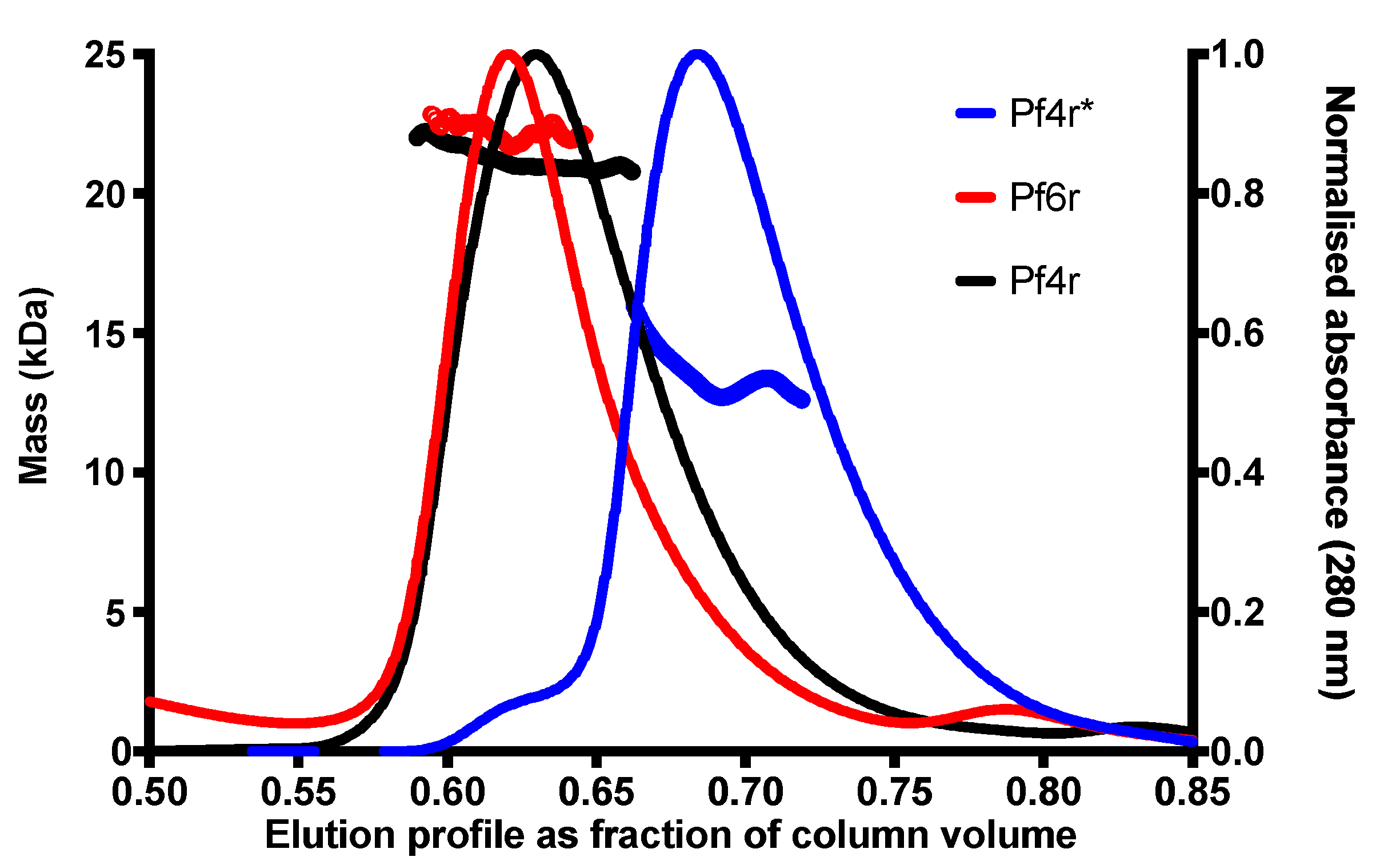
2.7. SEC-MALS Analysis of Purified Pf4r and Pf6r Proteins
2.8. Crystal Structure of the Pf4r* Protein
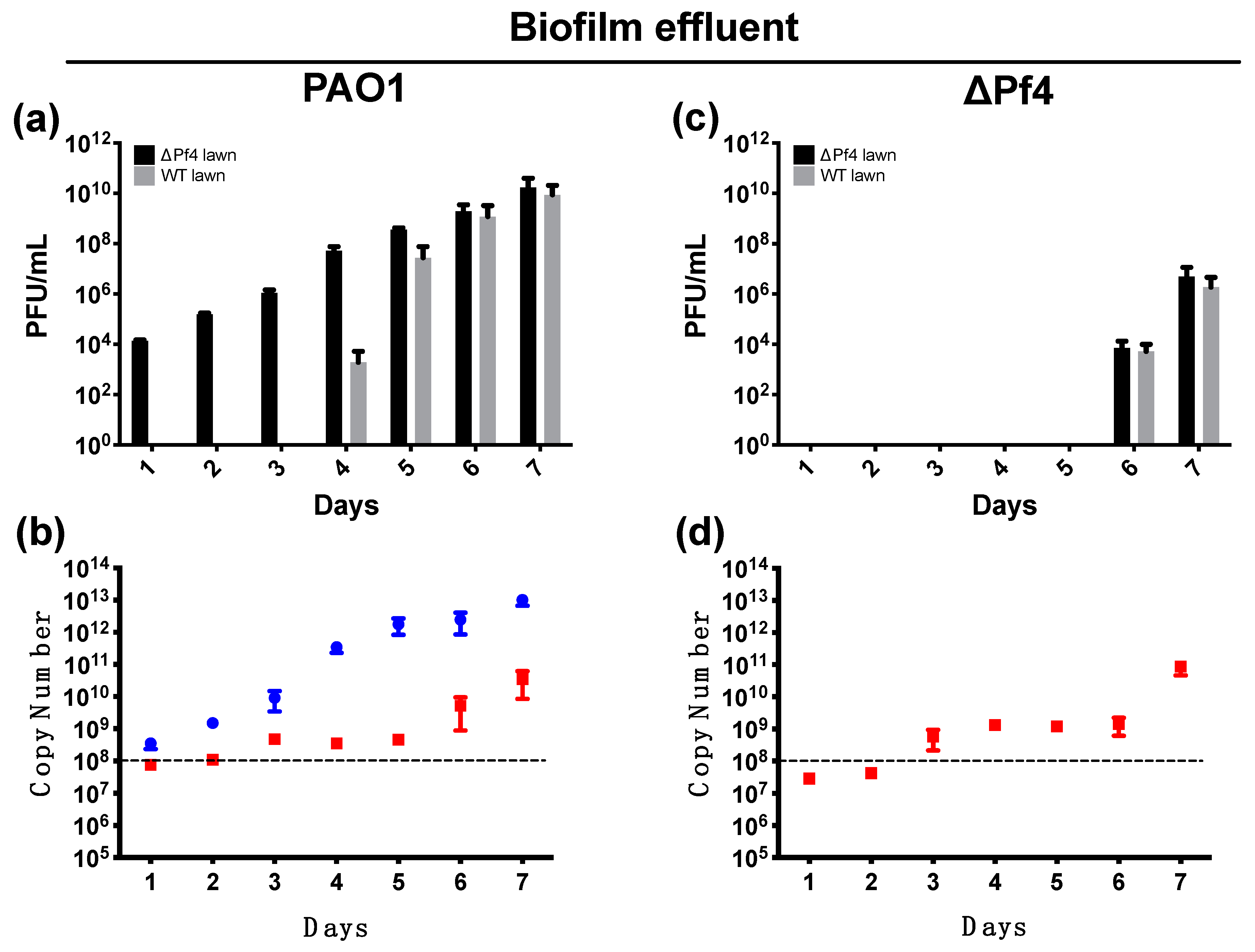
2.9. Tubing Biofilm System for Phenotypic Assays and mRNA Analysis
2.10. Pyoverdine and Pyocyanin Production
2.11. Production of Secreted Proteases
2.12. Estimation of Replication Fidelity and H2O2-Induced Mutant Frequencies
2.13. Construction and Expression of C-Terminal Hexahistidine Tagged Pf4r
2.14. In Vitro Immunity Assay of Pf4r Protein
2.15. Quantification of pf4r Expression by qRT-PCR
2.16. Chromatin Immunoprecipitation Sequencing (ChIPseq) Sample Preparation
2.17. ChIPseq Analysis
2.18. Transcriptomic Analysis of Pf4r Complemented Biofilms
3. Results
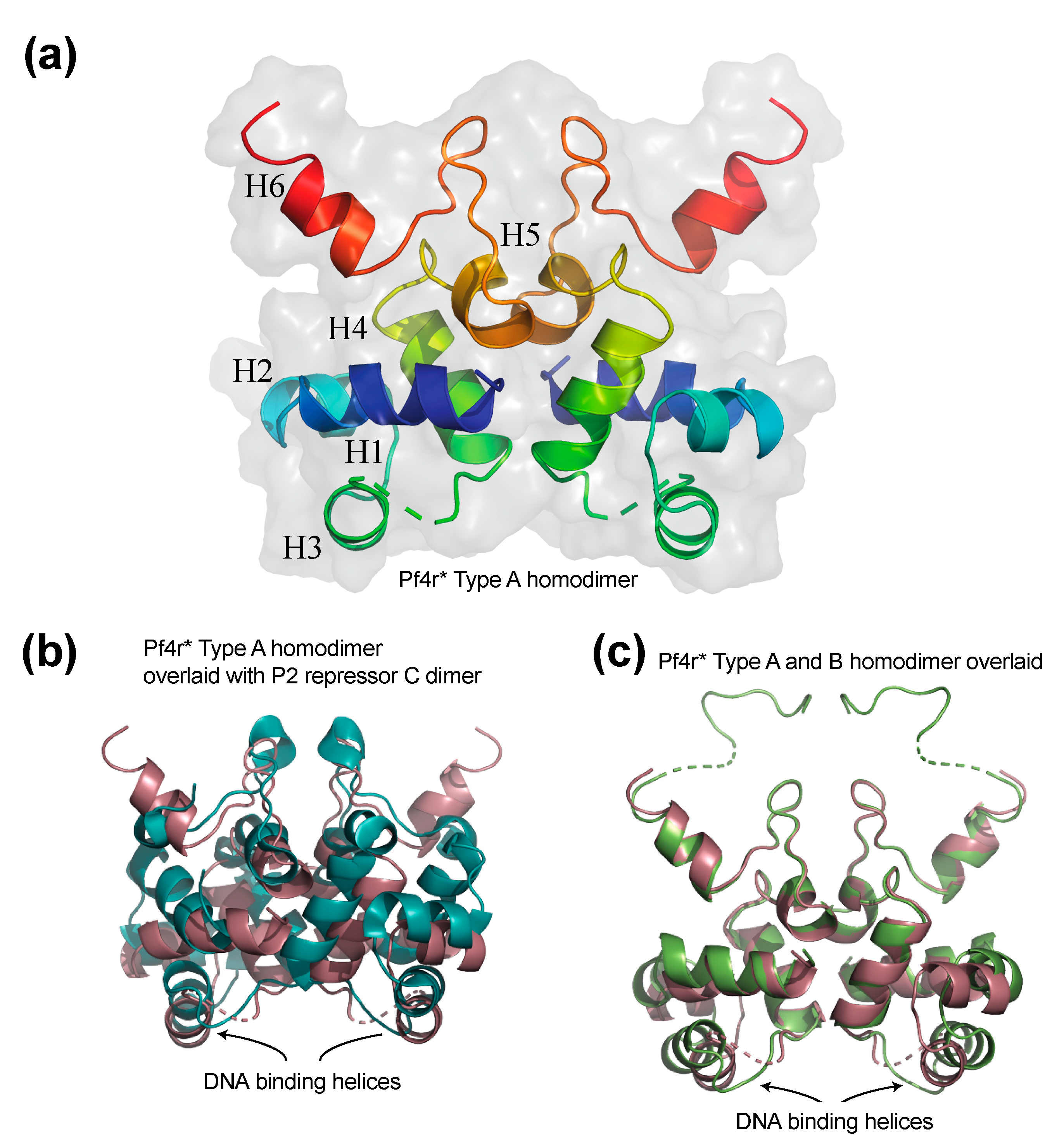
3.1. The Structure of the Pf4r Protein
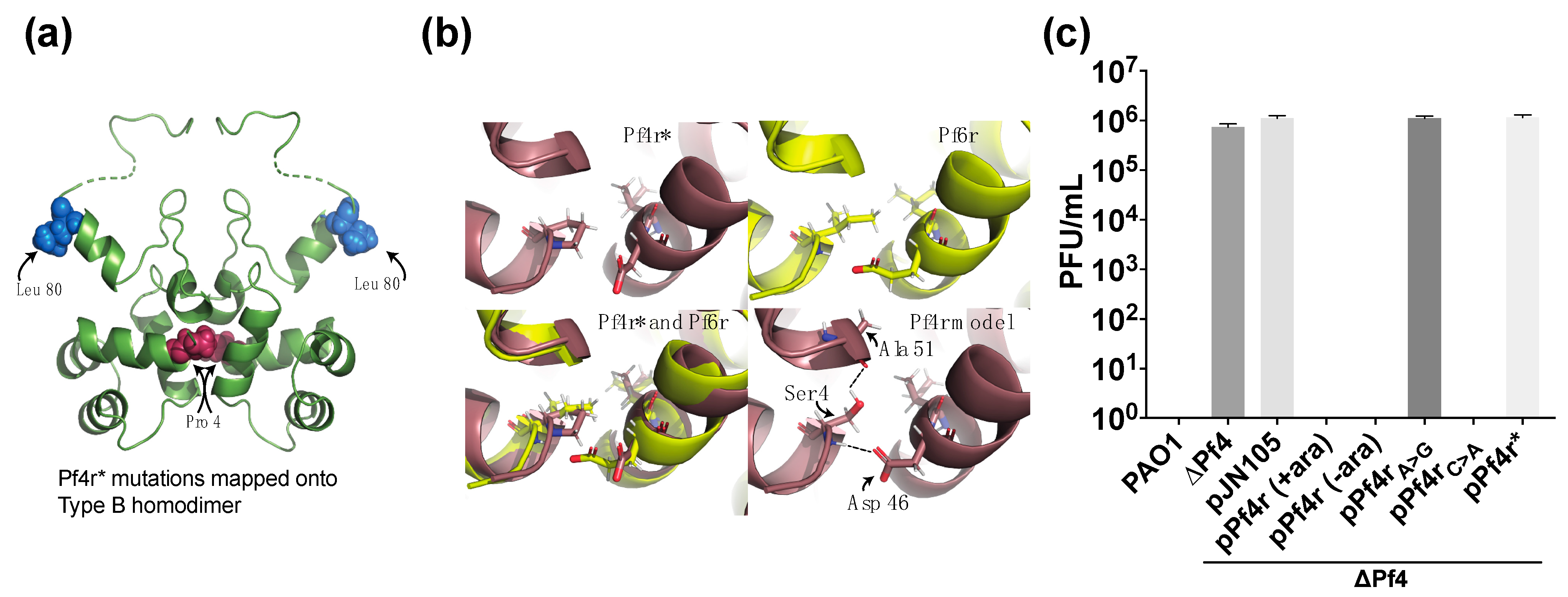
3.2. Mutations in the Dimerisation Interface Causes Loss of Immunity Function
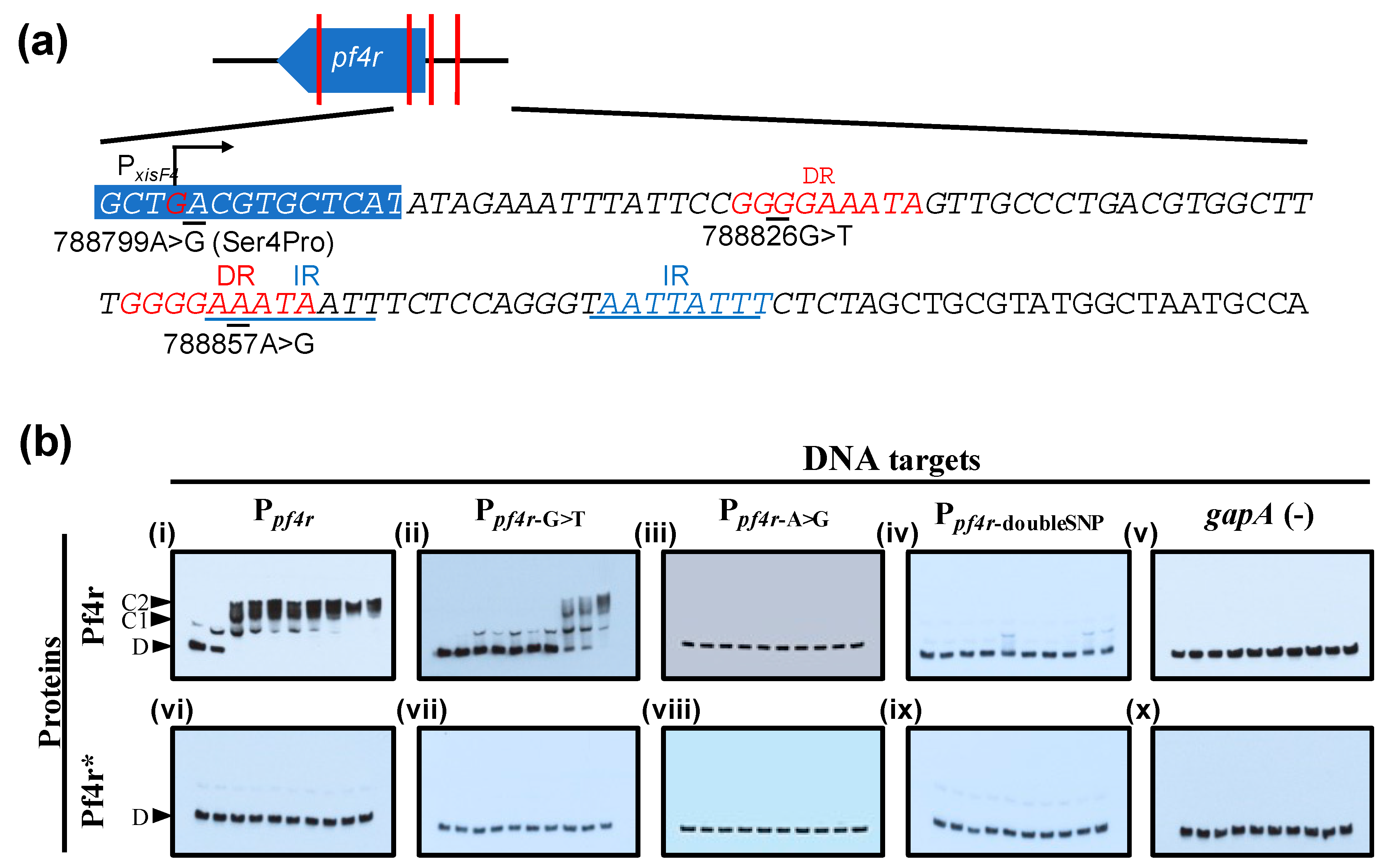
3.3. Mutations in the Promoter Regions and in Pf4r Affects DNA–Protein Binding
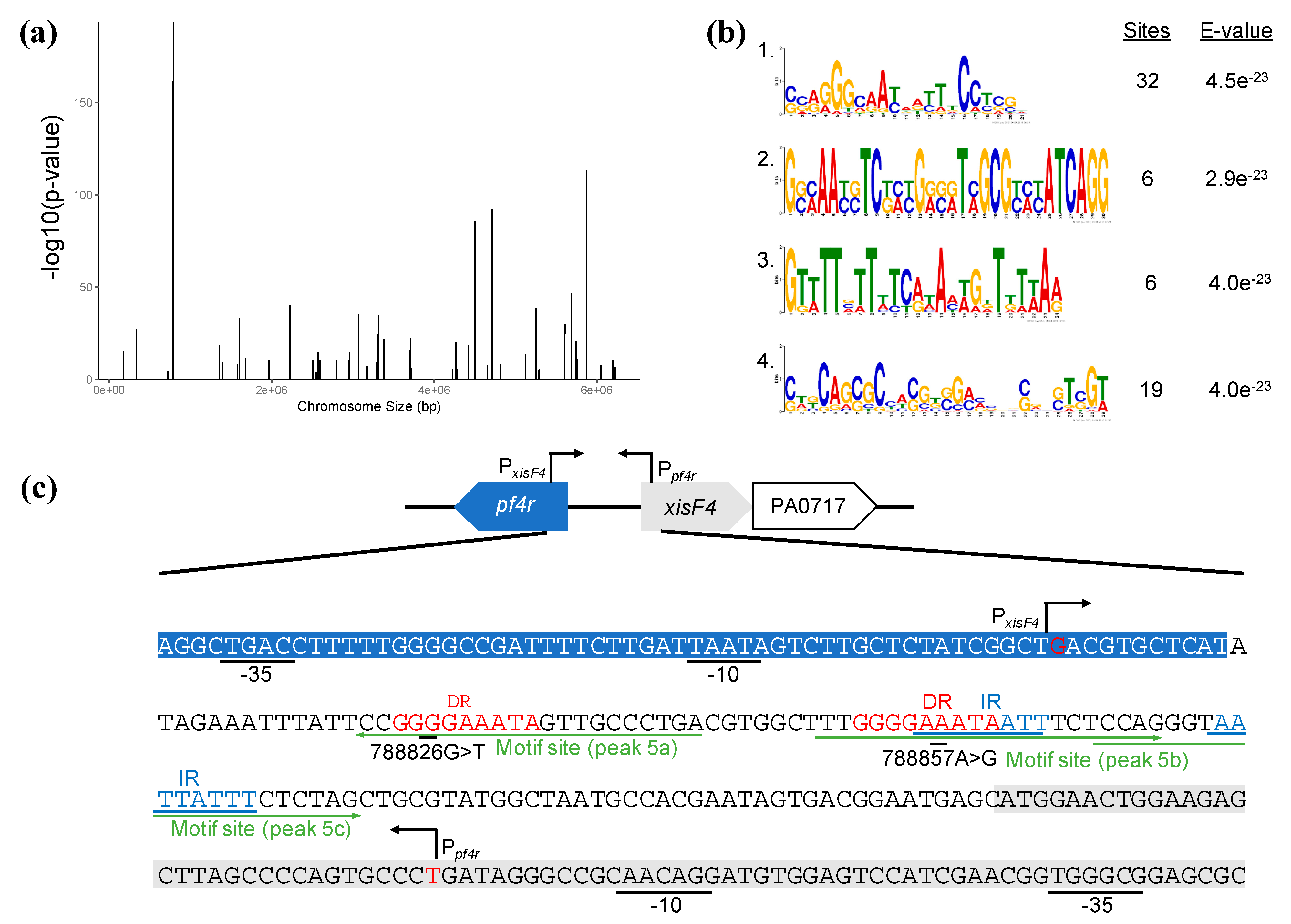
3.4. Pf4r Can Bind and Regulate Pseudomonas Genes
4. Discussion
4.1. Pf4r Binds to Conserved Promoter Sites as a Dimer
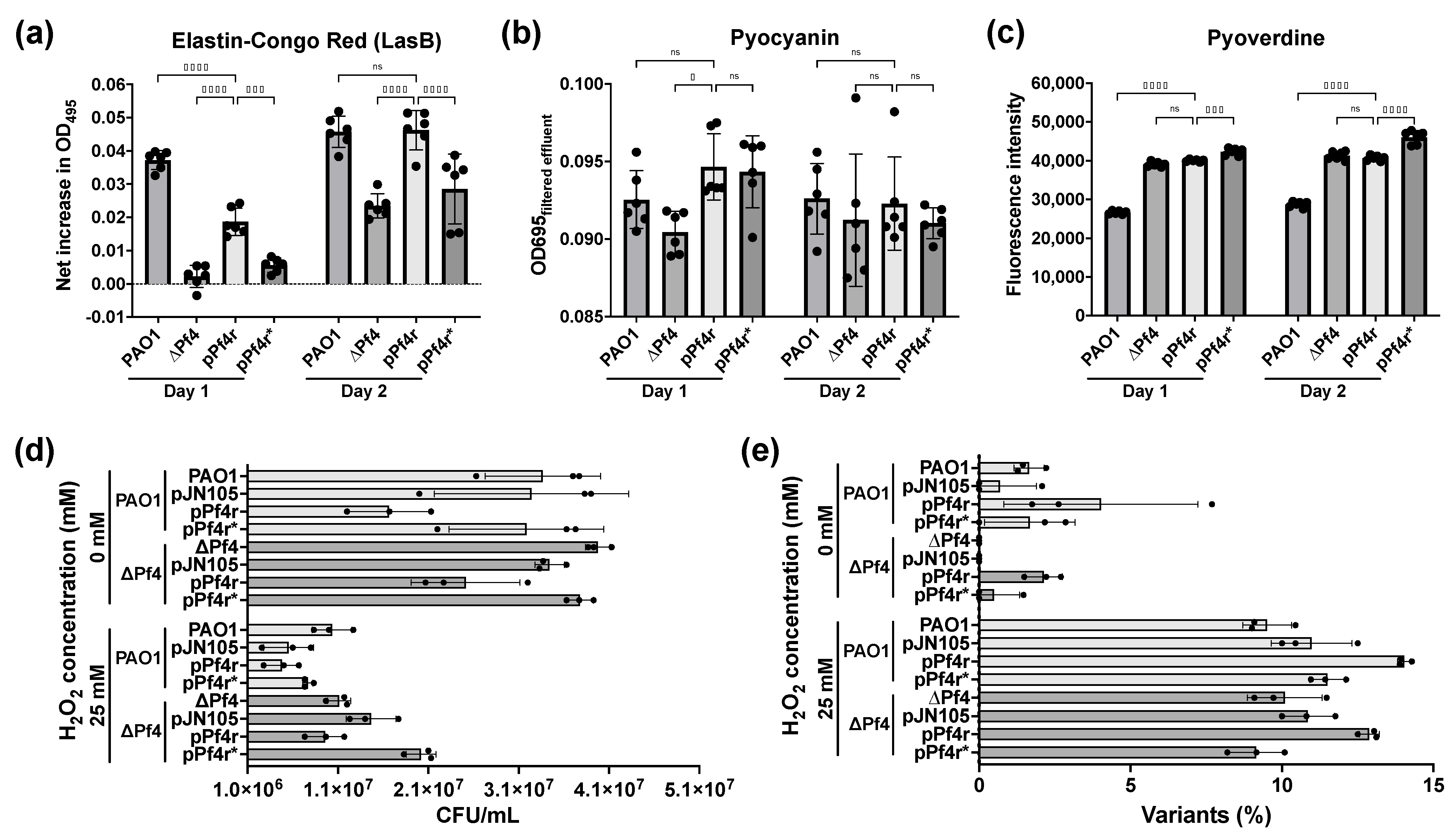
4.2. Pf4r Mediated Regulation of Host Genes Associated with Biofilm Development, Virulence and Mediating Superinfection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holmes, R.K. Biology and Molecular Epidemiology of Diphtheria Toxin and the tox Gene. J. Infect. Dis. 2000, 181, S156–S167. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Haarmann, N.; Schwidder, M.; Muniesa, M.; Schmidt, H. Bacteriophages of Shiga Toxin-Producing Escherichia coli and Their Contribution to Pathogenicity. Pathogens 2021, 10, 404. [Google Scholar] [CrossRef]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.K.; Waldor, M.K.; Friedman, D. Role for a Phage Promoter in Shiga Toxin 2 Expression from a Pathogenic Escherichia coli Strain. J. Bacteriol. 2001, 183, 2081–2085. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Singh, P.K. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. USA 2008, 105, 12503–12508. [Google Scholar] [CrossRef]
- Farrant, J.L.; Sansone, A.; Canvin, J.R.; Pallen, M.J.; Langford, P.R.; Wallis, T.S.; Dougan, G.; Kroll, J.S. Bacterial copper- and zinc-cofactored superoxide dismutase contributes to the pathogenesis of systemic salmonellosis. Mol. Microbiol. 1997, 25, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.S.; Ephang, K.K.S.; Etan, A.H.-M.; Eli, S.F.-Y.; Ow, D.S.-W. Small Colony Variants and Single Nucleotide Variations in Pf1 Region of PB1 Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2016, 7, 282. [Google Scholar] [CrossRef] [PubMed]
- Addy, H.S.; Askora, A.; Kawasaki, T.; Fujie, M.; Yamada, T. Loss of Virulence of the Phytopathogen Ralstonia solanacearum Through Infection by φRSM Filamentous Phages. Phytopathology 2012, 102, 469–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Argov, T.; Azulay, G.; Pasechnek, A.; Stadnyuk, O.; Sapir, S.R.; Borovok, I.; Sigal, N.; Herskovits, A.A. Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol. 2017, 38, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Genet. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.; Nayfach, S.; Schulz, F.; Sharrar, A.; Carnevali, P.B.M.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. ‘Big things in small packages: The genetics of filamentous phage and effects on fitness of their host’. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Munder, A.; Neugebauer, J.; Davenport, C.; Stanke, F.; Larbig, K.D.; Heeb, S.; Schöck, U.; Pohl, T.M.; Wiehlmann, L.; et al. Genome Diversity of Pseudomonas aeruginosa PAO1 Laboratory Strains. J. Bacteriol. 2010, 192, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Tan, C.H.; Mikkelsen, P.J.; Kung, V.; Woo, J.; Tay, M.; Hauser, A.; McDougald, D.; Webb, J.; Kjelleberg, S. The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J. 2008, 3, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.G.K.; Mai-Prochnow, A.; Kjelleberg, S.; McDougald, D.; Rice, S.A. Environmental cues and genes involved in establishment of the superinfective Pf4 phage of Pseudomonas aeruginosa. Front. Microbiol. 2014, 5, 654. [Google Scholar] [CrossRef]
- Secor, P.R.; Sweere, J.; Michaels, L.A.; Malkovskiy, A.V.; Lazzareschi, D.; Katznelson, E.; Rajadas, J.; Birnbaum, M.; Arrigoni, A.; Braun, K.R.; et al. Filamentous Bacteriophage Promote Biofilm Assembly and Function. Cell Host Microbe 2015, 18, 549–559. [Google Scholar] [CrossRef]
- Secor, P.R.; Burgener, E.B.; Kinnersley, M.; Jennings, L.K.; Roman-Cruz, V.; Popescu, M.; Van Belleghem, J.D.; Haddock, N.; Copeland, C.; Michaels, L.A.; et al. Pf Bacteriophage and Their Impact on Pseudomonas Virulence, Mammalian Immunity, and Chronic Infections. Front. Immunol. 2020, 11, 244. [Google Scholar] [CrossRef]
- Webb, J.; Lau, M.; Kjelleberg, S. Bacteriophage and Phenotypic Variation in Pseudomonas aeruginosa Biofilm Development. J. Bacteriol. 2004, 186, 8066–8073. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophages and Biofilms. In Biofilms: Formation, Development and Properties; Nova Science Publishers: Hauppauge, NY, USA, 2011; p. 58. [Google Scholar]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef]
- Tay, M. The Role of Bacteriophage in Granulation; Nanyang Technological University: Singapore, 2013. [Google Scholar]
- Li, Y.; Liu, X.; Tang, K.; Wang, P.; Zeng, Z.; Guo, Y.; Wang, X. Excisionase in Pf filamentous prophage controls lysis-lysogeny decision-making in Pseudomonas aeruginosa. Mol. Microbiol. 2018, 111, 495–513. [Google Scholar] [CrossRef]
- Hernandez-Doria, J.D.; Sperandio, V. Bacteriophage Transcription Factor Cro Regulates Virulence Gene Expression in Enterohemorrhagic Escherichia coli. Cell Host Microbe 2018, 23, 607–617. [Google Scholar] [CrossRef]
- Sambrook, J. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Hamlet in Long Island, NY, USA, 2001. [Google Scholar]
- Ismail, M.H. The Role of Bacteriophages in Mixed Microbial Communities and Populations of Pseudomonas Aeruginosa; Nanyang Technological University: Singapore, 2019. [Google Scholar]
- Some, D.; Amartely, H.; Tsadok, A.; Lebendiker, M. Characterization of Proteins by Size-Exclusion Chromatography Coupled to Multi-Angle Light Scattering (SEC-MALS). J. Vis. Exp. 2019, e59615. [Google Scholar] [CrossRef]
- Liebschner, D. Protein Crystallography—Methods and Protocols. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 74–75. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Adams, P.; Read, R.; McCoy, A.J.; Moriarty, N.W.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Zwart, P.H.; Hung, L.-W. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSolwizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 582–601. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Bunkóczi, G.; Echols, N.; McCoy, A.J.; Oeffner, R.; Adams, P.; Read, R.J. Phaser. MRage: Automated molecular replacement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Protein Interfaces, Surfaces and Assemblies’ Service PISA at European Bioinformatics Institute. 2007. Available online: http://www.ebi.ac.uk/pdbe/prot_int/pistart.html (accessed on 17 July 2019).
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.K.; Yam, J.K.H.; Mukherjee, M.; Periasamy, S.; Steinberg, P.D.; Kjelleberg, S.; Rice, S.A. Interspecific diversity reduces and functionally substitutes for intraspecific variation in biofilm communities. ISME J. 2015, 10, 846–857. [Google Scholar] [CrossRef]
- Kessler, E.; Safrin, M. Elastinolytic and Proteolytic Enzymes. In Pseudomonas Methods and Protocols; Filloux, A., Ramos, J.-L., Eds.; Springer: New York, NY, USA, 2014; pp. 135–169. [Google Scholar]
- Rodríguez-Rojas, A.; Blazquez, J. The Pseudomonas aeruginosa pfpI Gene Plays an Antimutator Role and Provides General Stress Protection. J. Bacteriol. 2009, 191, 844–850. [Google Scholar] [CrossRef]
- Webb, J.S.; Thompson, L.S.; James, S.; Charlton, T.; Tolker-Nielsen, T.; Koch, B.; Givskov, M.; Kjelleberg, S. Cell Death in Pseudomonas aeruginosa Biofilm Development. J. Bacteriol. 2003, 185, 4585–4592. [Google Scholar] [CrossRef] [PubMed]
- Bonocora, R.P.; Wade, J.T. ChIP-Seq for Genome-Scale Analysis of Bacterial DNA-Binding Proteins. Methods Mol. Biol. 2015, 1276, 327–340. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2015. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 23 January 2019).
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137–R139. [Google Scholar] [CrossRef] [PubMed]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Münch, R.; Hiller, K.; Grote, A.; Scheer, M.; Klein, J.; Schobert, M.; Jahn, D. Virtual Footprint and PRODORIC: An integrative framework for regulon prediction in prokaryotes. Bioinformatics 2005, 21, 4187–4189. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McElroy, K.E.; Hui, J.G.K.; Woo, J.K.K.; Luk, A.W.S.; Webb, J.; Kjelleberg, S.; Rice, S.A.; Thomas, T. Strain-specific parallel evolution drives short-term diversification during Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. USA 2014, 111, E1419–E1427. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 2.4.
- Massad, T.; Skaar, K.; Nilsson, H.; Damberg, P.; Henriksson-Peltola, P.; Haggård-Ljungquist, E.; Högbom, M.; Stenmark, P. Crystal structure of the P2 C-repressor: A binder of non-palindromic direct DNA repeats. Nucleic Acids Res. 2010, 38, 7778–7790. [Google Scholar] [CrossRef]
- Maiti, R.; Van Domselaar, G.H.; Zhang, H.; Wishart, D.S. SuperPose: A simple server for sophisticated structural superposition. Nucleic Acids Res. 2004, 32, W590–W594. [Google Scholar] [CrossRef]
- Meisner, J.; Goldberg, J.B. The Escherichia coli rhaSR-PrhaBAD Inducible Promoter System Allows Tightly Controlled Gene Expression over a Wide Range in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2016, 82, 6715–6727. [Google Scholar] [CrossRef] [PubMed]
- Alonso, B.; Fernández-Barat, L.; Di Domenico, E.G.; Marín, M.; Cercenado, E.; Merino, I.; De Pablos, M.; Muñoz, P.; Guembe, M. Characterization of the virulence of Pseudomonas aeruginosa strains causing ventilator-associated pneumonia. BMC Infect. Dis. 2020, 20, 1–8. [Google Scholar] [CrossRef]
- Berngruber, T.W.; Weissing, F.J.; Gandon, S. Inhibition of Superinfection and the Evolution of Viral Latency. J. Virol. 2010, 84, 10200–10208. [Google Scholar] [CrossRef]
- Bednarz, M.; Halliday, J.A.; Herman, C.; Golding, I. Revisiting Bistability in the Lysis/Lysogeny Circuit of Bacteriophage Lambda. PLoS ONE 2014, 9, e100876. [Google Scholar] [CrossRef]
- Fernández, L.; Breidenstein, E.B.M.; Song, D.; Hancock, R. Role of Intracellular Proteases in the Antibiotic Resistance, Motility, and Biofilm Formation of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 56, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in Bacteriophage Lambda Development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef]
- Ptashne, M. A Genetic Switch, Third Edition, Phage Lambda Revisited; Cold Spring Harbor Laboratory Press: Hamlet in Long Island, NY, USA, 2004. [Google Scholar]
- Addy, H.S.; Askora, A.; Kawasaki, T.; Fujie, M.; Yamada, T. The Filamentous Phage ϕRSS1 Enhances Virulence of Phytopathogenic Ralstonia solanacearum on Tomato. Phytopathology 2012, 102, 244–251. [Google Scholar] [CrossRef]
- Cheng, C.-M.; Wang, H.-J.; Bau, H.-J.; Kuo, T.-T. The primary immunity determinant in modulating the lysogenic immunity of the filamentous bacteriophage cf. J. Mol. Biol. 1999, 287, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Quinones, M.; Kimsey, H.H.; Waldor, M.K. LexA Cleavage Is Required for CTX Prophage Induction. Mol. Cell 2005, 17, 291–300. [Google Scholar] [CrossRef]
- Kimsey, H.H.; Waldor, M.K. The CTXϕ Repressor RstR Binds DNA Cooperatively to Form Tetrameric Repressor-Operator Complexes. J. Biol. Chem. 2004, 279, 2640–2647. [Google Scholar] [CrossRef]
- Hay, I.D.; Lithgow, T. Filamentous phages: Masters of a microbial sharing economy. EMBO Rep. 2019, 20, e47427. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Gene | ChIPseq (MACS2) | Binding Motif (Virtual Footprint) | Gene Product | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Peak | Fold Enrichment | −log10 p Value | Conserved Motif | Strand | Distance from Gene Start | PWM | Location | |||
| PA0154 | pcaG | peak_1 | 2.50081 | 15.46022 | CGAGGGCCACTATTACTTCCG | + | 309 | 10.40 | coding region | protocatechuate 3,4-dioxygenase, ⍺ subunit |
| PA0299 | spuC | peak_2 | 3.20044 | 27.26002 | CGAGGGCAACAAGATCCTCGA | + | - | 10.68 | coding region | polyamine:pyruvate transaminase |
| PA0717 | PA0717 | peak_5a | 6.37126 | 179.65939 | TCAGGGCAACTATTTCCCCGG | - | 302 | 10.92 | intergenic | hypothetical protein of bacteriophage Pf1 |
| peak_5b | 1.9989 | 7.87121 | TTGGGGAAATAATTTCTCCAG | + | 274 | 9.87 | intergenic | hypothetical protein of bacteriophage Pf1 | ||
| peak_5c | 1.84109 | 6.05157 | CCAGGGTAATTATTTCTCTAG | + | 257 | 10.48 | intergenic | hypothetical protein of bacteriophage Pf1 | ||
| PA1249 | aprA | peak_6 | 2.83107 | 18.79315 | CCAGGCGAACAATTGCCCGCA | - | 324 | 10.39 | intergenic | alkaline metalloproteinase precursor |
| PA1450 | PA1450 | peak_8a | 1.59375 | 3.78181 | CAAGGGCAACGATTTCCTGGC | + | 314 | 11.62 | coding region | conserved hypothetical protein |
| peak_8b | 1.69336 | 4.73467 | conserved hypothetical protein | |||||||
| PA1477 | ccmC | peak_9 | 3.58171 | 33.07233 | GCAGGGCAATAGCTTCCGCAT | + | - | 10.90 | coding region | heme exporter protein CcmC |
| PA1538 | PA1538 | peak_10 | 2.33997 | 11.48559 | CCAGGGCAACCATTACACCGC | - | - | 10.72 | coding region | probable flavin-containing monooxygenase |
| PA1806 | fabI | peak_11 | 2.28219 | 10.71977 | GCAGGGCAAGTACAACCTCAC | - | 206 | 9.79 | coding region | NADH-dependent enoyl-ACP reductase |
| PA2035 | PA2035 | peak_12 | 3.95772 | 40.04172 | TAGAGGAAATAGTTACATAGA | - | 246 | 10.64 | intergenic | probable decarboxylase |
| PA2306 | ambA | peak_14 | 1.63948 | 3.92643 | CCAGAGCAACAGTTGCCGAGA | + | 57 | 9.78 | intergenic | putative LysE-type translocator |
| PA2327 | PA2327 | peak_15 | 2.57107 | 14.75297 | CGAAGGCAATAATGCCTTCCT | - | 136 | 10.81 | coding region | probable permease of ABC transporter |
| PA2345 | PA2345 | peak_16 | 2.28219 | 10.71977 | CCAGGGCAACAGTTACCGGCG | - | 108 | 10.73 | intergenic | conserved hypothetical protein |
| PA2475 | PA2475 | peak_17 | 2.26436 | 10.55864 | CCAGGGCAGCAGTTGCCCGGT | + | - | 9.68 | coding region | probable cytochrome P450 |
| PA2612 | serS | peak_18 | 2.57107 | 14.75297 | CGGAGAGGATTATTTCCCCGA | - | 246 | 9.98 | coding region | seryl-tRNA synthetase |
| PA2715 | PA2715 | peak_19 | 3.72661 | 35.24215 | GCAGGGAAATATTGCCCGGCT | - | 193 | 10.29 | coding region | probable ferredoxin |
| GCCGGGCAATATTTCCCTGCC | + | 192 | 11.07 | coding region | ||||||
| PA2953 | fixC | peak_22 | 3.49856 | 34.68851 | CGAGGGCAACTATATCATCTC | + | - | 10.70 | coding region | electron transfer flavoprotein-ubiquinone oxidoreductase |
| PA3316 | PA3316 | peak_25 | 1.83515 | 6.34533 | CCGGGGAAATGATTTCCTACA | + | 194 | 11.67 | coding region | probable permease of ABC transporter |
| PA3766 | PA3766 | peak_26 | 1.80342 | 5.48047 | CAAGGGCGAGAAGTTCATCAG | - | - | 10.02 | coding region | probable aromatic amino acid transporter |
| TCATGACAACAACTCCCTGCA | + | - | 9.77 | coding region | ||||||
| PA4021 | PA4021 | peak_30 | 5.40382 | 85.6179 | CGAGGGCTGCTATTTCCTCCG | - | - | 9.78 | coding region | probable transcriptional regulator |
| PA4210 | phzA1 | peak_32a | 5.29093 | 85.06443 | CCGGAGAAACTTTTCCCTCGC | - | 32 | 9.76 | intergenic | probable phenazine biosynthesis protein |
| peak_32b | 1.9649 | 6.99041 | CCGGAGAAACTTTTCCCTCGC | |||||||
| PA4293 | pprA | peak_33 | 2.07667 | 8.56197 | CCGGGGCAACGTTTTCTCTGG | - | - | 9.61 | coding region | two-component sensor PprA |
| PA4576 | PA4576 | peak_34 | 2.45922 | 13.79064 | CCAGGGTGATGGTTTCCTCGT | + | - | 11.44 | coding region | probable ATP-dependent protease |
| PA4704 | cbpA | peak_37 | 1.78182 | 5.43821 | GGGAGTAAATATCTTCCCGTG | - | 73 | 9.73 | intergenic | cAMP-binding protein A |
| GCAGGCGGATCGGTTCTTCGT | - | - | 9.64 | coding region | ||||||
| PA5040 | pilQ | peak_39 | 3.86006 | 46.60398 | GCAGGGCAATATCACCCTGCG | - | - | 10.49 | coding region | Type 4 fimbrial biogenesis outer membrane protein PilQ precursor |
| GCAGGGTGATATTGCCCTGCA | + | - | 10.3 | coding region | ||||||
| CGAGGGCAACGTGGTCATCGA | - | - | 9.91 | coding region | ||||||
| GCGGGTCAAGCAGTTCCTGGA | - | 295 | 9.84 | coding region | ||||||
| PA5089 | tle5b | peak_40 | 2.82994 | 20.5095 | GCGGGTGAATATCTGCCCCCA | + | - | 9.82 | coding region | type VI secretion phospholipase D effector Tle5b |
| PA5103 | puuR | peak_41 | 2.26708 | 10.92837 | GCAAGGAAATCGCTACCCCGT | + | 273 | 10.44 | coding region | probable transcriptional regulator |
| PA5205 | PA5205 | peak_42 | 6.52198 | 113.39552 | GCGGGGTGATCTTTTCCTCGT | - | - | 10.89 | coding region | conserved hypothetical protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, M.H.; Michie, K.A.; Goh, Y.F.; Noorian, P.; Kjelleberg, S.; Duggin, I.G.; McDougald, D.; Rice, S.A. The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression. Viruses 2021, 13, 1614. https://doi.org/10.3390/v13081614
Ismail MH, Michie KA, Goh YF, Noorian P, Kjelleberg S, Duggin IG, McDougald D, Rice SA. The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression. Viruses. 2021; 13(8):1614. https://doi.org/10.3390/v13081614
Chicago/Turabian StyleIsmail, Muhammad Hafiz, Katharine A. Michie, Yu Fen Goh, Parisa Noorian, Staffan Kjelleberg, Iain G. Duggin, Diane McDougald, and Scott A. Rice. 2021. "The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression" Viruses 13, no. 8: 1614. https://doi.org/10.3390/v13081614
APA StyleIsmail, M. H., Michie, K. A., Goh, Y. F., Noorian, P., Kjelleberg, S., Duggin, I. G., McDougald, D., & Rice, S. A. (2021). The Repressor C Protein, Pf4r, Controls Superinfection of Pseudomonas aeruginosa PAO1 by the Pf4 Filamentous Phage and Regulates Host Gene Expression. Viruses, 13(8), 1614. https://doi.org/10.3390/v13081614