
Genomic Epidemiology and Evolution of Duck Hepatitis A Virus


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and Sequencing
2.2. Sequence Analysis
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hisham, I.; Ellakany, H.F.; Selim, A.A.; Abdalla, M.A.M.; El-Abideen, M.A.Z.; Kilany, W.H.; Ali, A.; Elbestawy, A.R. Comparative pathogenicity of duck hepatitis A virus-1 isolates in experimentally infected Pekin and Muscovy ducklings. Front. Vet. Sci. 2020, 7, 234. [Google Scholar] [CrossRef]
- Levine, P.P.; Fabricant, J. A hitherto-undescribed virus disease of ducks in North America. Cornell Vet. 1950, 40, 71–86. [Google Scholar]
- Liu, R.; Shi, S.; Huang, Y.; Chen, Z.; Chen, C.; Cheng, L.; Fu, G.; Chen, H.; Wan, C.; Fu, Q. Comparative pathogenicity of different subtypes of duck hepatitis A virus in Pekin ducklings. Vet. Microbiol. 2019, 228, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Ma, H.; Ding, Y.; Li, Z.; Sun, Y.; Li, M.; Shi, Y. The pathogenicity of duck hepatitis A virus types 1 and 3 on ducklings. Poult. Sci. 2019, 98, 6333–6339. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Liu, B.; Sun, C.; Zhao, L.; Chen, H. Construction of the recombinant duck enteritis virus delivering capsid protein VP0 of the duck hepatitis A virus. Vet. Microbiol. 2020, 249, 108837. [Google Scholar] [CrossRef]
- Kim, M.C.; Kwon, Y.K.; Joh, S.J.; Lindberg, A.M.; Kwon, J.H.; Kim, J.H.; Kim, S.J. Molecular analysis of duck hepatitis virus type 1 reveals a novel lineage close to the genus Parechovirus in the family Picornaviridae. J. Gen. Virol. 2006, 87, 3307–3316. [Google Scholar] [CrossRef]
- Tseng, C.H.; Knowles, N.J.; Tsai, H.J. Molecular analysis of duck hepatitis virus type 1 indicates that it should be assigned to a new genus. Virus Res. 2007, 123, 190–203. [Google Scholar] [CrossRef]
- Hassan, T.I.R.; Eid, A.A.M.; Ghanem, I.A.I.; Shahin, A.M.; Adael, S.A.A.; Mohamed, F.F. First report of duck hepatitis A virus 3 from duckling flocks of Egypt. Avian Dis. 2020, 64, 269–276. [Google Scholar] [CrossRef]
- Kim, M.C.; Kwon, Y.K.; Joh, S.J.; Kim, S.J.; Tolf, C.; Kim, J.H.; Sung, H.W.; Lindberg, A.M.; Kwon, J.H. Recent Korean isolates of duck hepatitis virus reveal the presence of a new geno- and serotype when compared to duck hepatitis virus type 1 type strains. Arch. Virol. 2007, 152, 2059–2072. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tsai, H.J. Molecular characterization of a new serotype of duck hepatitis virus. Virus Res. 2007, 126, 19–31. [Google Scholar] [CrossRef]
- The Picornavirus Page. Available online: https://www.picornaviridae.com/sg4/avihepatovirus/avihepatovirus_a/dhav-1_seq.htm (accessed on 14 May 2021).
- Doan, H.T.T.; Le, X.T.K.; Do, R.T.; Hoang, C.T.M.; Nguyen, K.T.; Le, T.H. Molecular genotyping of duck hepatitis A viruses (DHAV) in Vietnam. J. Infect. Dev. Ctries. 2016, 10, 988–995. [Google Scholar] [CrossRef]
- Kamomae, M.; Kameyama, M.; Ishii, J.; Nabe, M.; Ogura, Y.; Iseki, H.; Yamamoto, Y.; Mase, M. An outbreak of duck hepatitis A virus type 1 infection in Japan. J. Vet. Med. Sci. 2017, 79, 917–920. [Google Scholar] [CrossRef]
- Kozdruń, W.; Czekaj, H.; Lorek, M. Outbreak of duck viral hepatitis in duckling flocks in Poland. Bull. Vet. Inst. Pulawy 2014, 58, 513–515. [Google Scholar] [CrossRef][Green Version]
- Kaleta, E.F. Duck viral hepatitis type 1 vaccination: Monitoring of the immune response with a microneutralisation test in Pekin duck embryo kidney cell cultures. Avian Pathol. 1988, 17, 325–332. [Google Scholar] [CrossRef]
- MacPherson, L.W.; Avery, R.J. Duck virus hepatitis in Canada. Can. J. Comp. Med. Vet. Sci. 1957, 21, 26–31. [Google Scholar]
- Nikita, N.V.; Leonov, I.K.; Yavdoshak, L.I. Interferonogenic activity of the strain BH-3 duck hepatitis virus type I (Picornaviridae: Avihepatovirus: Avihepatovirus A). Vopr. Virusol. 2021, 66, 162–166. (In Russian) [Google Scholar] [CrossRef]
- Palya, V.; Ivanics, E.; Glavits, R.; Skare, J.; Nagy, E.; Bakonyi, T.; Bajusz, I. Reoccurrence of duck viral hepatitis epidemics—possibilities for prevention and control. Magy Állatorvosok Lapja 2006, 128, 281–287. (In Hungarian) [Google Scholar]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst. Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucl. Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Molecular Evolution, Phylogenetics and Epidemiology. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 14 May 2021).
- Liu, G.; Wang, F.; Ni, Z.; Yun, T.; Yu, B.; Huang, J.; Chen, J. Genetic diversity of the VP1 gene of duck hepatitis virus type I (DHV-I) isolates from Southeast China is related to isolate attenuation. Virus Res. 2008, 137, 137–141. [Google Scholar] [CrossRef]
- Ma, X.; Sheng, Z.; Huang, B.; Qi, L.; Li, Y.; Yu, K.; Liu, C.; Qin, Z.; Wang, D.; Song, M.; et al. Molecular Evolution and Genetic Analysis of the Major Capsid Protein VP1 of Duck Hepatitis A Viruses: Implications for Antigenic Stability. PLoS ONE 2015, 10, e0132982. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, Y.; Lan, J.; Xie, Z.; Zhang, X.; Jiang, S. Evidence of possible vertical transmission of duck hepatitis A virus type 1 in ducks. Transbound. Emerg. Dis. 2020, 68, 267–275. [Google Scholar] [CrossRef]
- Kuroda, M.; Niwa, S.; Sekizuka, T.; Tsukagoshi, H.; Yokoyama, M.; Ryo, A.; Sato, H.; Kiyota, N.; Noda, M.; Kozawa, K.; et al. Molecular evolution of the VP1, VP2 and VP3 genes in human rhinovirus species C. Sci. Rep. 2015, 5, 8185. [Google Scholar] [CrossRef]
- Lu, L.; Dung, N.V.; Bryant, J.E.; Carrique-Mas, J.; Cuong, N.V.; Anh, P.H.; Rabaa, M.A.; Baker, S.; Simmonds, P.; Woolhouse, M.E. Evolution and phylogeographic dissemination of endemic porcine picornaviruses in Vietnam. Virus Evol. 2016, 2, vew001. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wen, X.; Zhu, D.; Cheng, A.; Wang, M.; Chen, S.; Jia, R.; Liu, M.; Sun, K.; Zhao, X.; Yang, Q.; et al. Molecular epidemiology of duck hepatitis a virus types 1 and 3 in China, 2010–2015. Transbound. Emerg. Dis. 2018, 65, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- The Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef]
- Kaszab, E.; Marton, S.; Dán, Á.; Farsang, A.; Bálint, Á.; Bányai, K.; Fehér, E. Molecular epidemiology and phylodynamics of goose haemorrhagic polyomavirus. Transbound. Emerg. Dis. 2020, 67, 2602–2608. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Zhang, Z.; Liu, S.; Zhang, Y. Goose haemorrhagic hepatitis caused by a new subtype duck hepatitis type 1 virus. Vet. Microbiol. 2011, 152, 280–283. [Google Scholar] [CrossRef]
- Shi, S.; Chen, H.; Chen, Z.; Fu, G.; Wan, C.; Huang, Y.; Lin, S.; Cheng, L.; Fu, Q.; Lin, J.; et al. Complete genome sequence of a duck hepatitis a virus 1 isolated from a pigeon in China. Genome Announc. 2013, 1, e00451-13. [Google Scholar] [CrossRef]






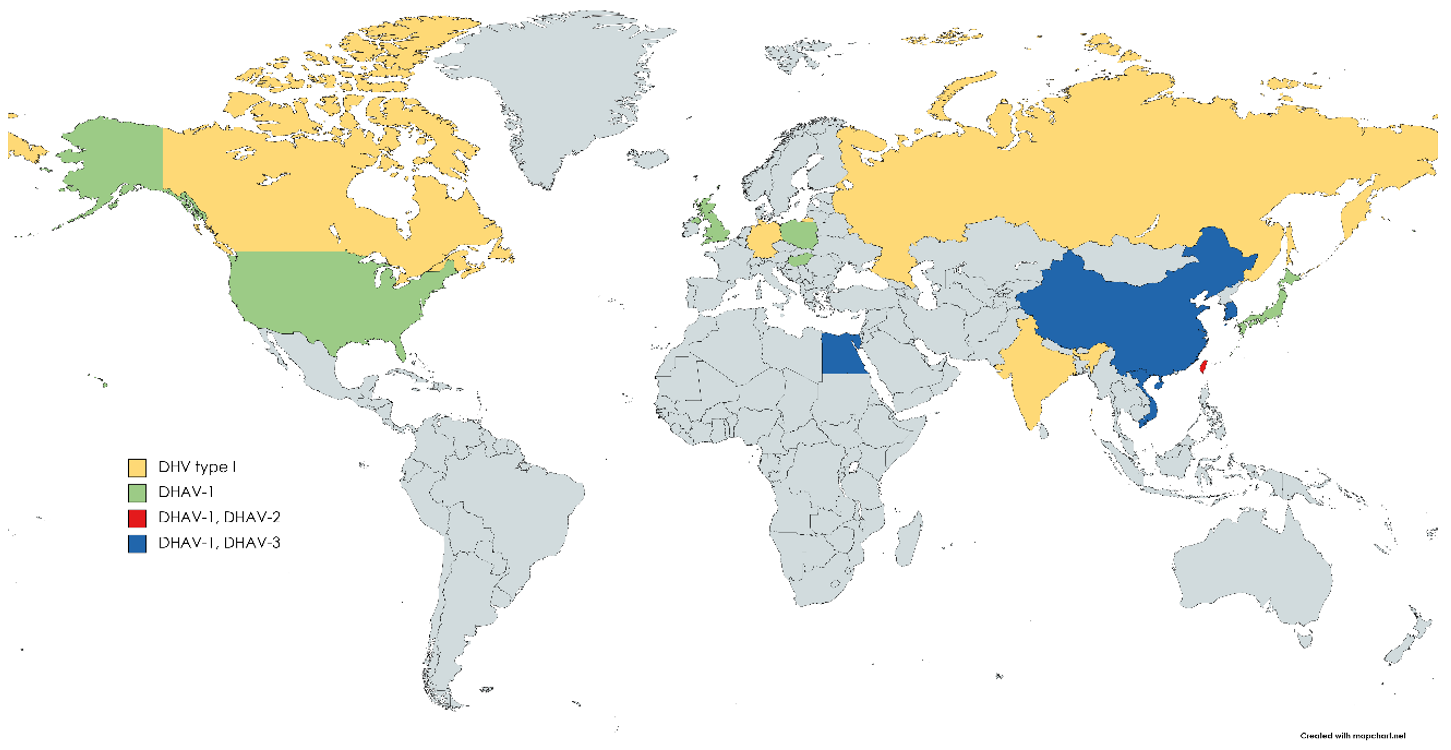{kind=link}
{kind=link}
{kind=link}
{kind=link}
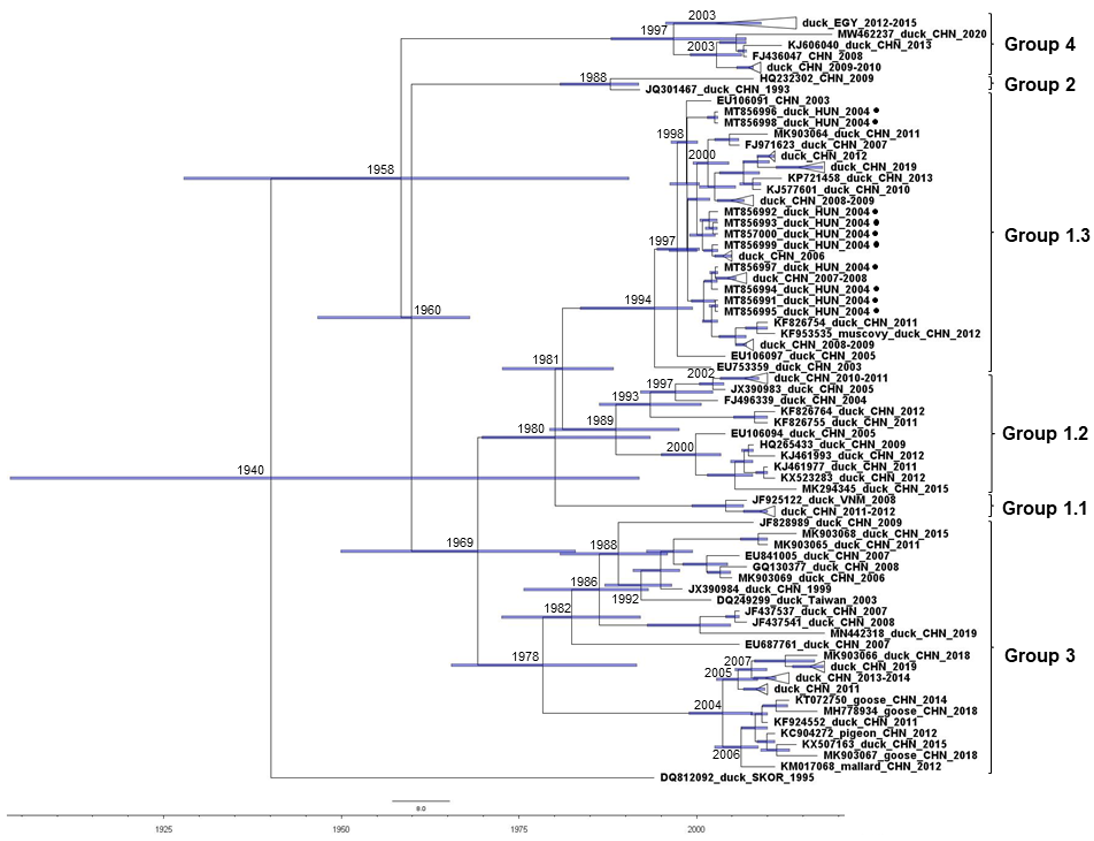{kind=link}
| Primer Name | Orientation | 5′–3′ Sequence |
|---|---|---|
| DHAV_F | forward | GGAGGTGGTGCTGAAA |
| DHAV_R1 | reverse | GTGGAAAGAAGAGAGCTGGC |
| DHAV_F2 | forward | GATGTGGCATGTTGTYAAYCGAC |
| DHAV_R2 | reverse | CAACCAAAYGCTGATGTTTTGG |
| DHAV_F3 | forward | CCAACYCTTCARTGGCTCCAG |
| DHAV_R3 | reverse | CATTTGGCTTAGGGTCCTCAC |
| Event * | Region | Recombinant | Major Parent | Minor Parent | Recombination Type | p-Values |
|---|---|---|---|---|---|---|
| #1 | VP0 | JF828994-996 ** | JX235698, KC993890, GQ485310 | JF828997 | Intergenotype | 10−11–10−176 |
| #2 | 3A-3D | JF828996 | KC893553 | JF828994 | Intragenotype | 10−8–10−16 |
| #3 | VP0 | EU888310 | MT856991 to MT857000 | DQ249299 | Intragenotype | 10−6–10−7 |
| Region | Model | dN/dS | Evolutionary Rate s/s/y | 95% HPD |
|---|---|---|---|---|
| VP0 | HKY + G | 0.0945 | 7.0037 × 10−4 | 2.3584 × 10−4 to 1.2087 × 10−3 |
| VP1 | HKY + G | 0.159 | 7.8508 × 10−4 | 3.1147 × 10−4 to 1.2961 × 10−3 |
| VP3 | HKY + G | 0.105 | 9.0959 × 10−4 | 3.6255 × 10−4 to 1.5174 × 10−3 |
| 2A1 | HKY | 0.087 | 1.1147 × 10−3 | 2.5081 × 10−4 to 2.2143 × 10−3 |
| 2A2 | HKY + G | 0.111 | 8.3308 × 10−4 | 3.0703 × 10−4 to 1.3598 × 10−3 |
| 2B | HKY + G | 0.0489 | 8.112 × 10−4 | 2.2148 × 10−4 to 1.4842 × 10−3 |
| 2C | HKY + G | 0.0472 | 6.1681 × 10−4 | 2.0083 × 10−4 to 1.0726 × 10−3 |
| 3A | GTR + G | 0.0545 | 8.1151 × 10−4 | 2.1100 × 10−4 to 1.5012 × 10−3 |
| 3B | HKY + G | 0.127 | 1.059 × 10−3 | 2.5708 × 10−4 to 1.9493 × 10−3 |
| 3C | HKY + G | 0.0466 | 6.8276 × 10−4 | 2.7878 × 10−4 to 1.1399 × 10−3 |
| 3D | HKY + G | 0.0719 | 5.6286 × 10−4 | 1.8270 × 10−4 to 9.9198 × 10−4 |
| VP1 extended sequence set | HKY + G + I | 0.191 | 1.013 × 10−3 | 6.7481 × 10−4 to 1.3458 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, E.; Jakab, S.; Bali, K.; Kaszab, E.; Nagy, B.; Ihász, K.; Bálint, Á.; Palya, V.; Bányai, K. Genomic Epidemiology and Evolution of Duck Hepatitis A Virus. Viruses 2021, 13, 1592. https://doi.org/10.3390/v13081592
Fehér E, Jakab S, Bali K, Kaszab E, Nagy B, Ihász K, Bálint Á, Palya V, Bányai K. Genomic Epidemiology and Evolution of Duck Hepatitis A Virus. Viruses. 2021; 13(8):1592. https://doi.org/10.3390/v13081592
Chicago/Turabian StyleFehér, Enikő, Szilvia Jakab, Krisztina Bali, Eszter Kaszab, Borbála Nagy, Katalin Ihász, Ádám Bálint, Vilmos Palya, and Krisztián Bányai. 2021. "Genomic Epidemiology and Evolution of Duck Hepatitis A Virus" Viruses 13, no. 8: 1592. https://doi.org/10.3390/v13081592
APA StyleFehér, E., Jakab, S., Bali, K., Kaszab, E., Nagy, B., Ihász, K., Bálint, Á., Palya, V., & Bányai, K. (2021). Genomic Epidemiology and Evolution of Duck Hepatitis A Virus. Viruses, 13(8), 1592. https://doi.org/10.3390/v13081592