
Active Components of Commonly Prescribed Medicines Affect Influenza A Virus–Host Cell Interaction: A Pilot Study

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds, Cells, and Viruses
2.2. Cell Viability Assay
2.3. Transcriptomics Analysis
2.4. Metabolomics Analysis
2.5. Bioinformatics Analysis
3. Results
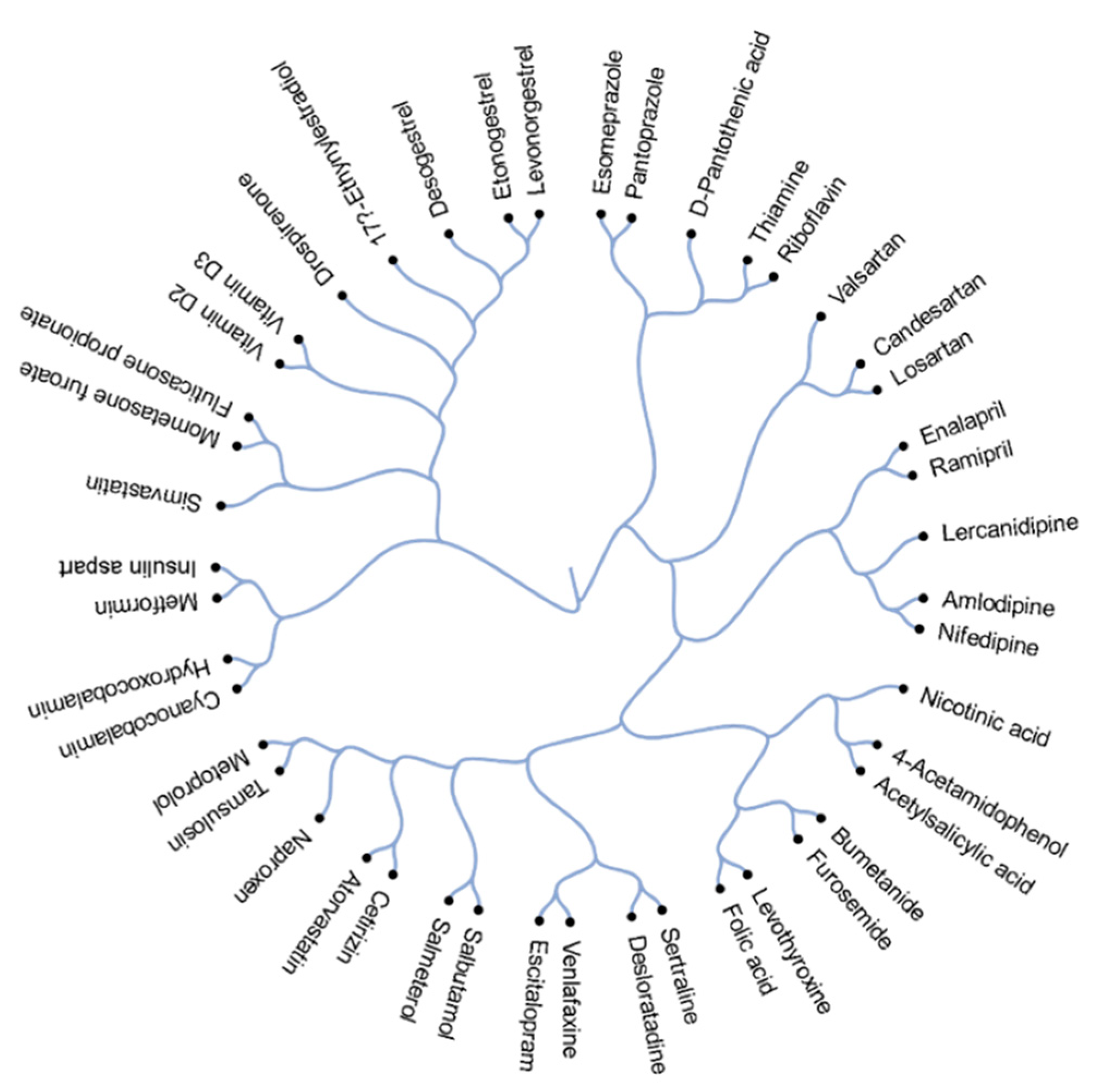
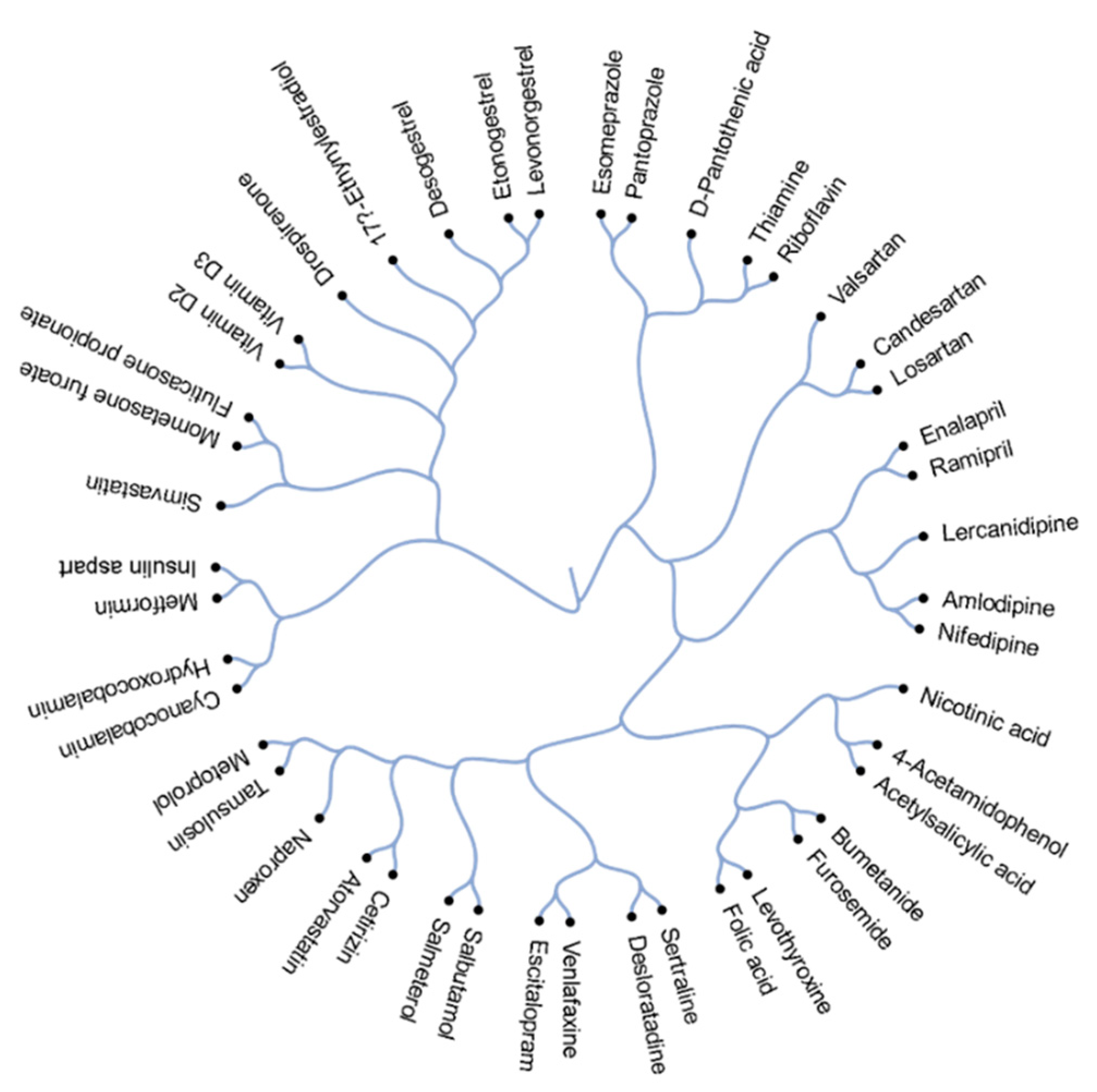
3.1. Structural Comparison of 45 Active Components of Commonly Prescribed Drugs
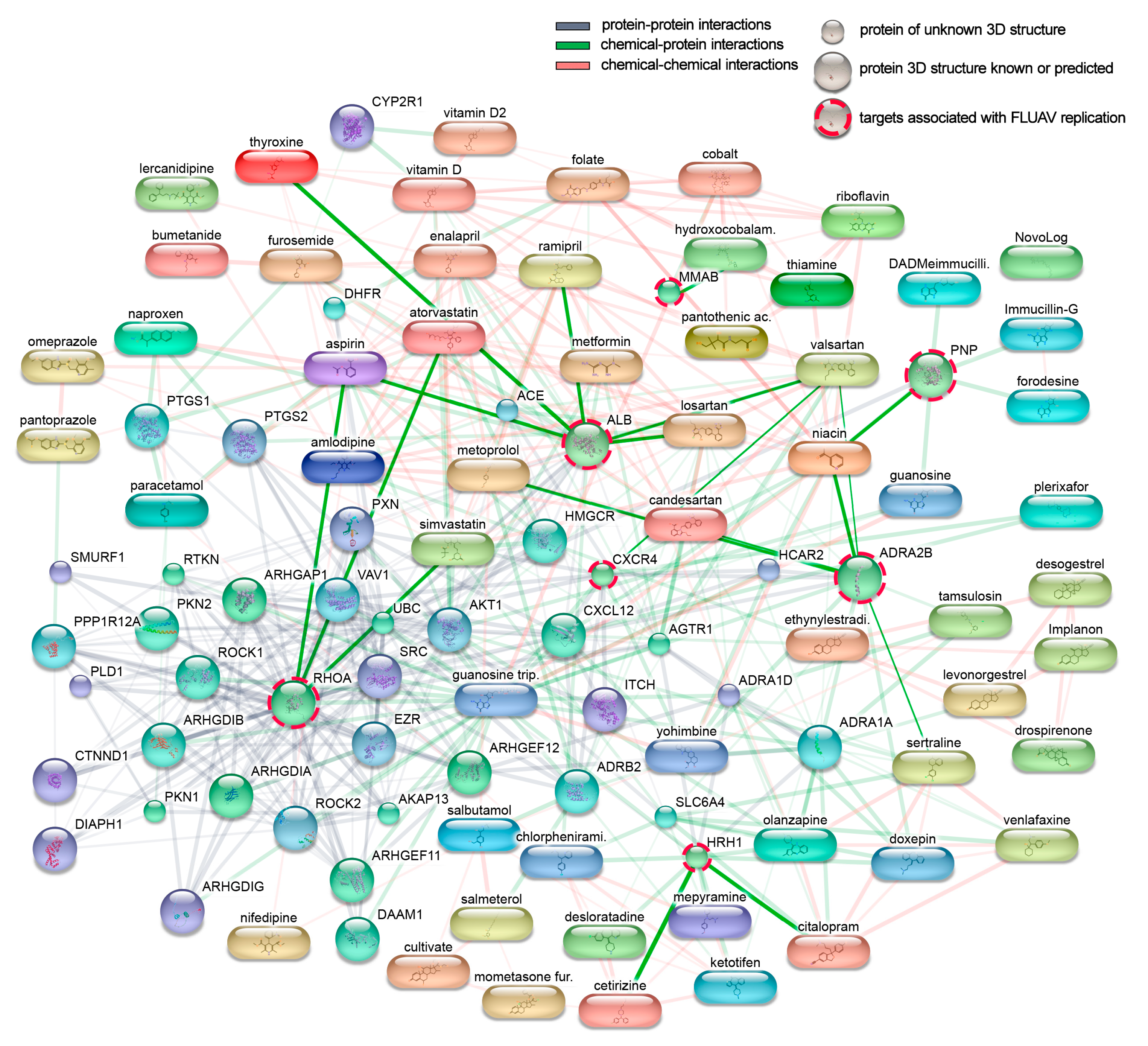
3.2. Cellular Targets of Active Components of The Most Prescribed Drugs and Their Potential Effect on FLUAV–Host Cell Interaction
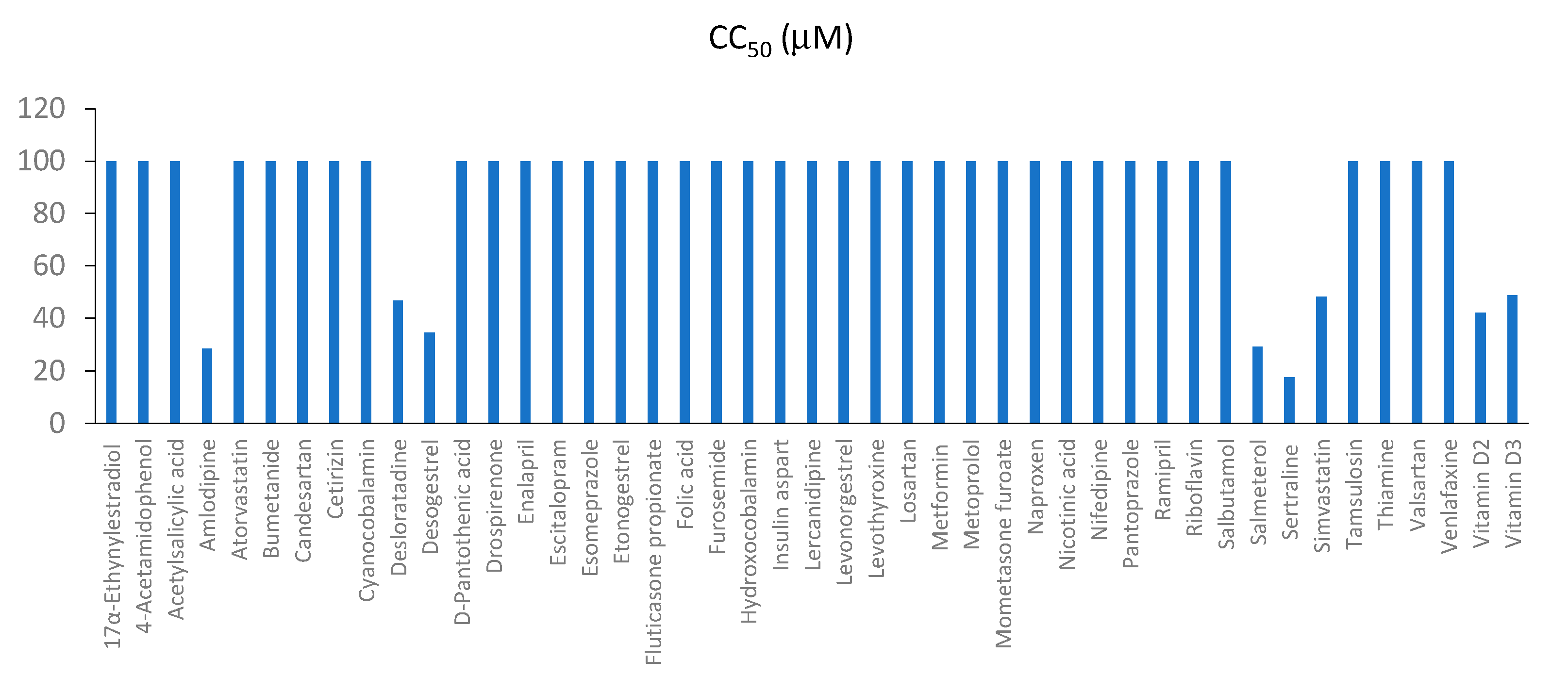
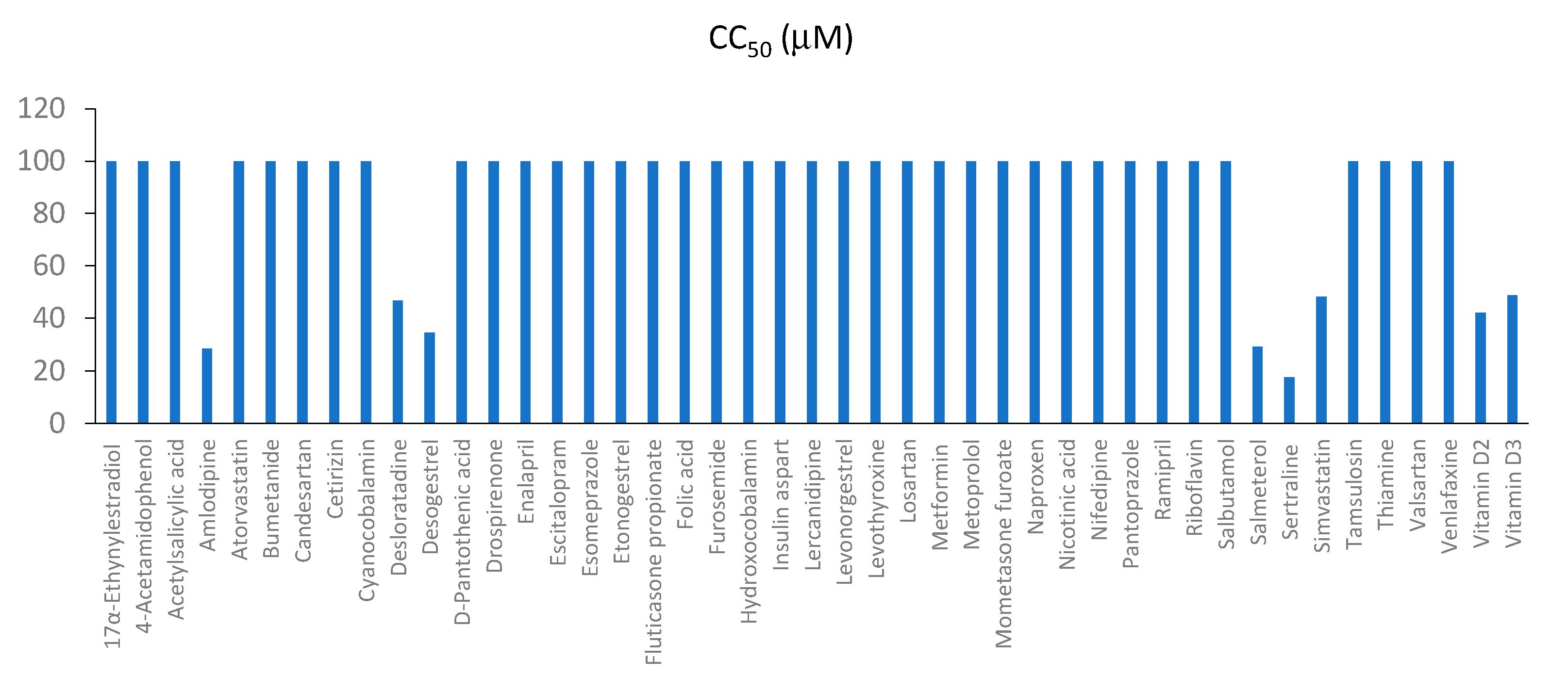
3.3. Toxicity and Efficacy of Active Components of Commonly Prescribed Medicines in Mock- and FLUAV-Infected RPE Cells
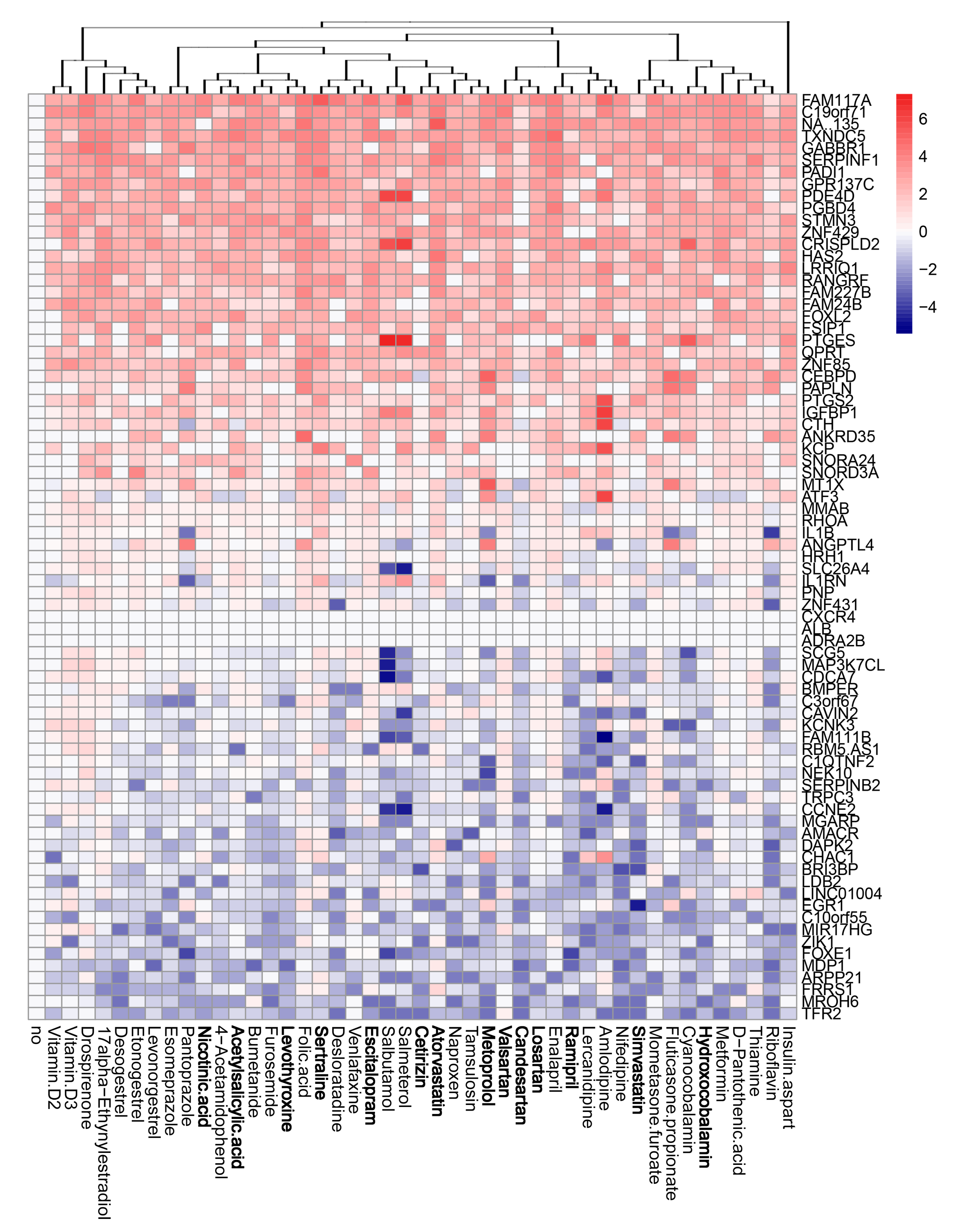
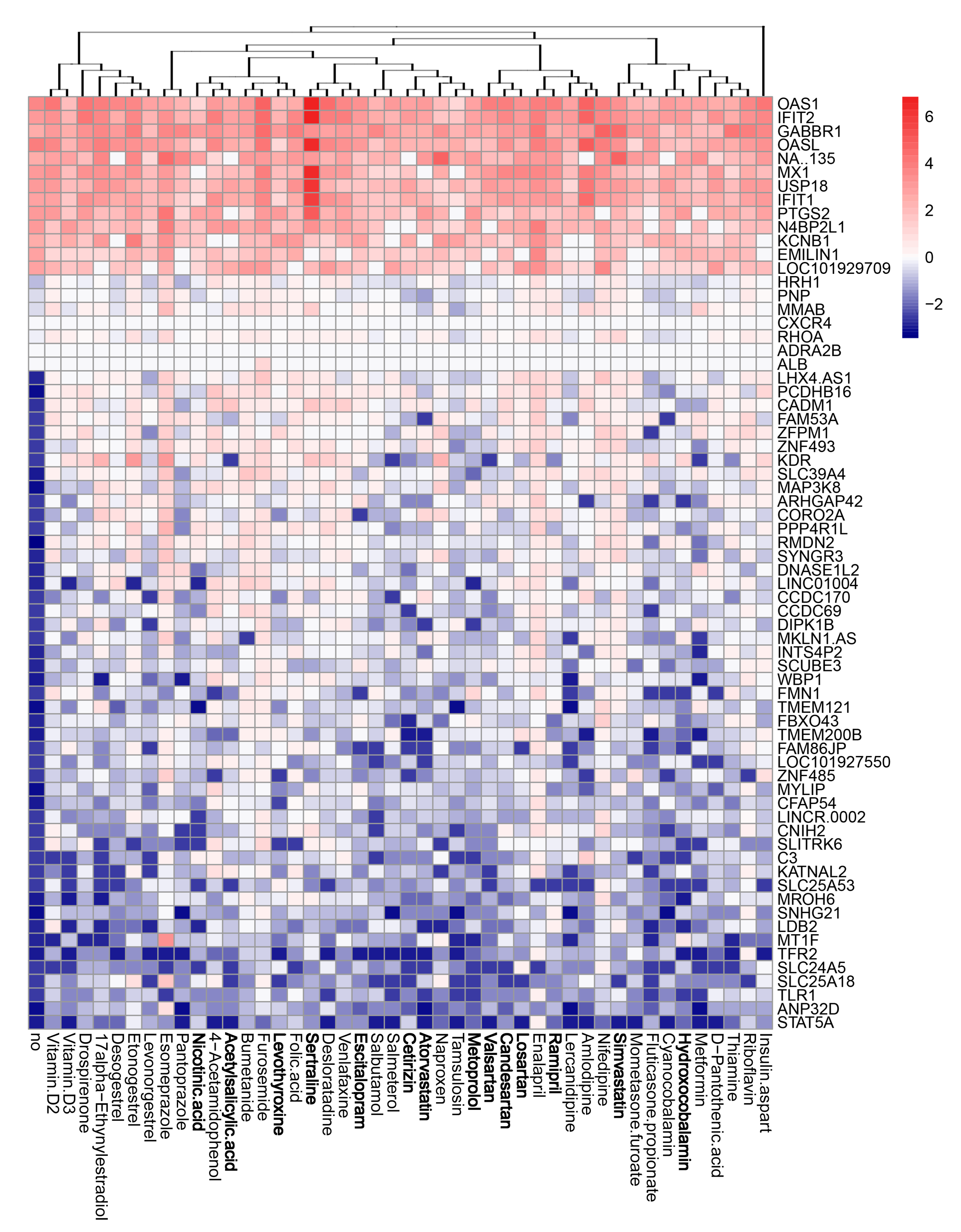
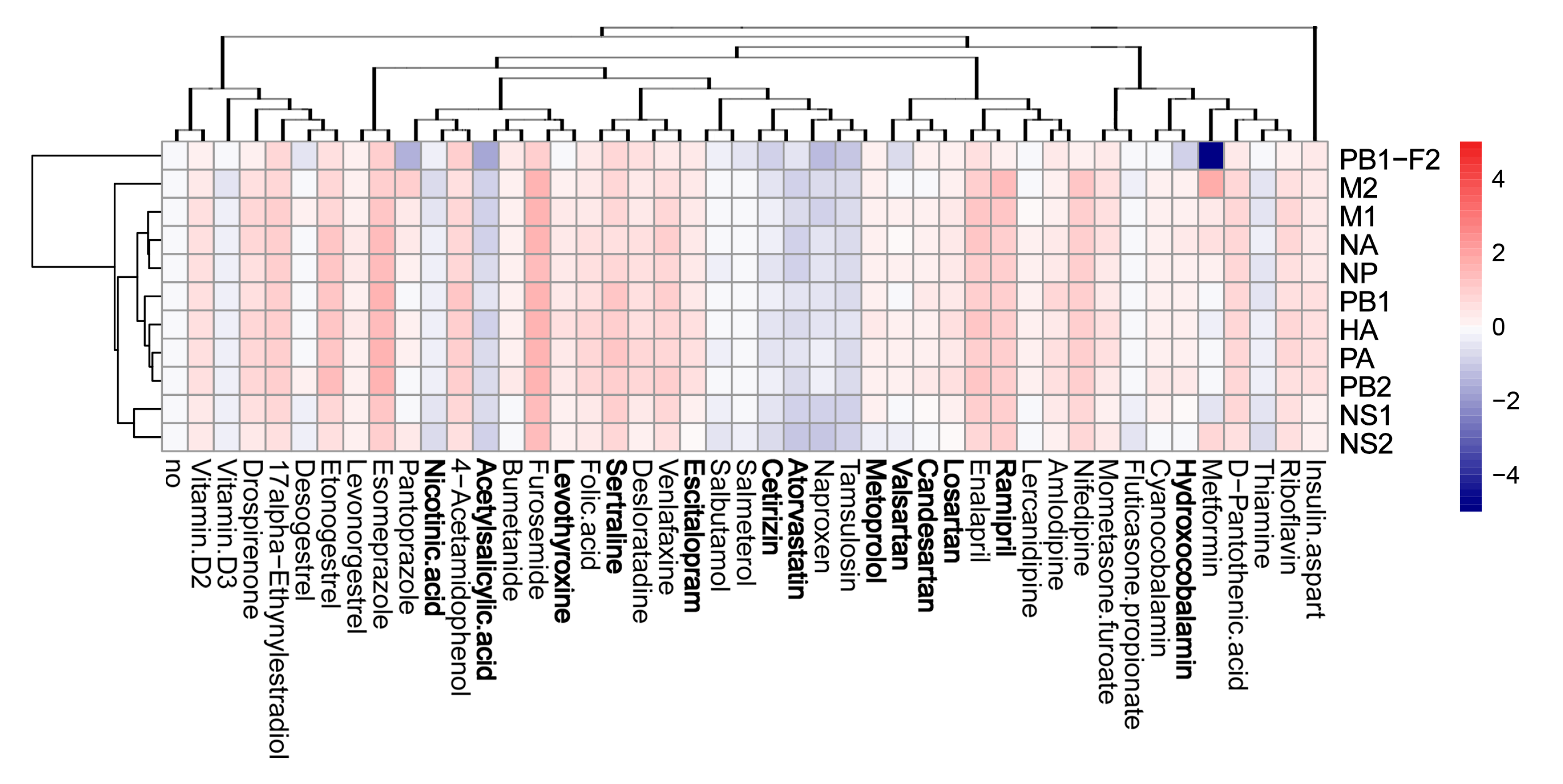
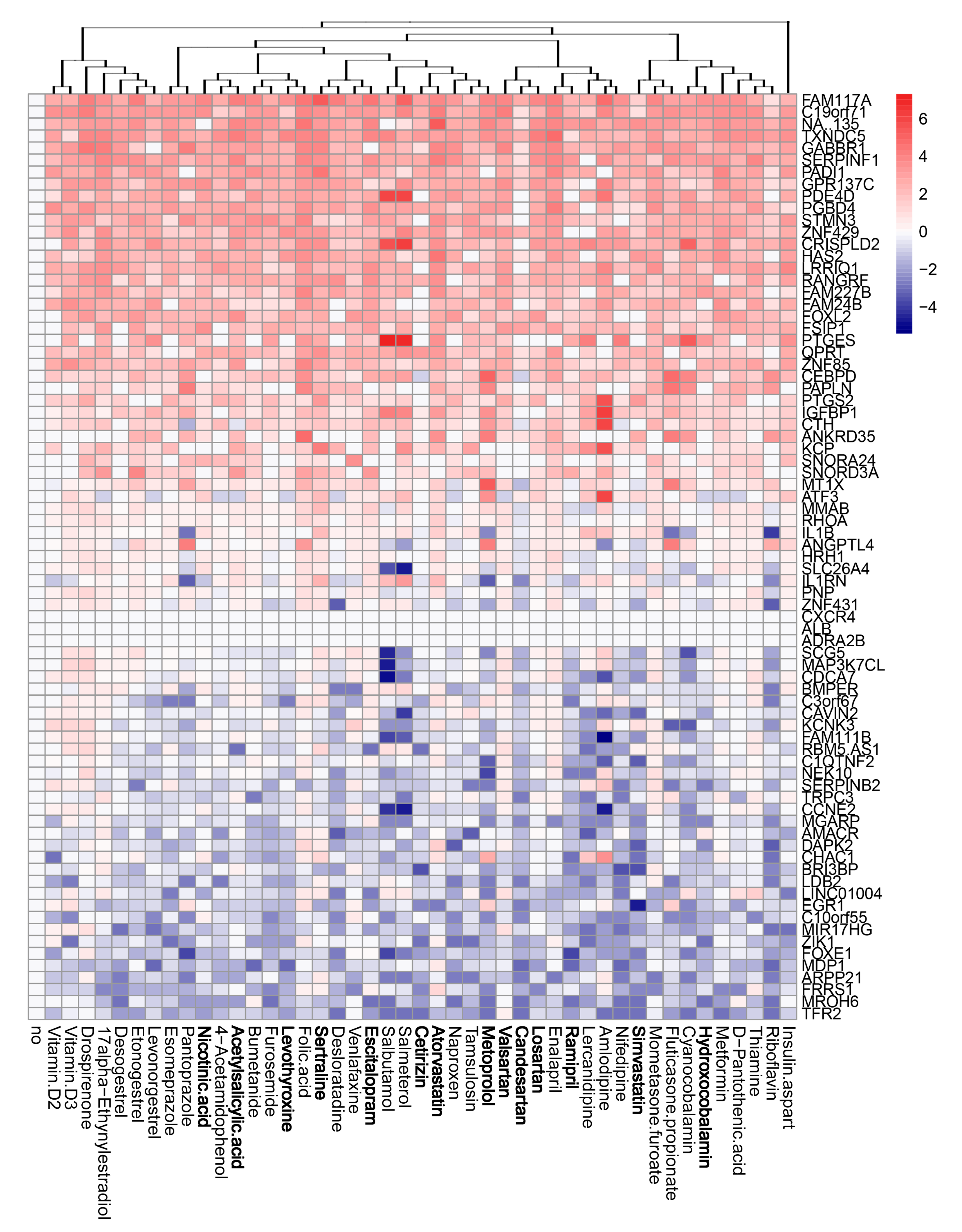
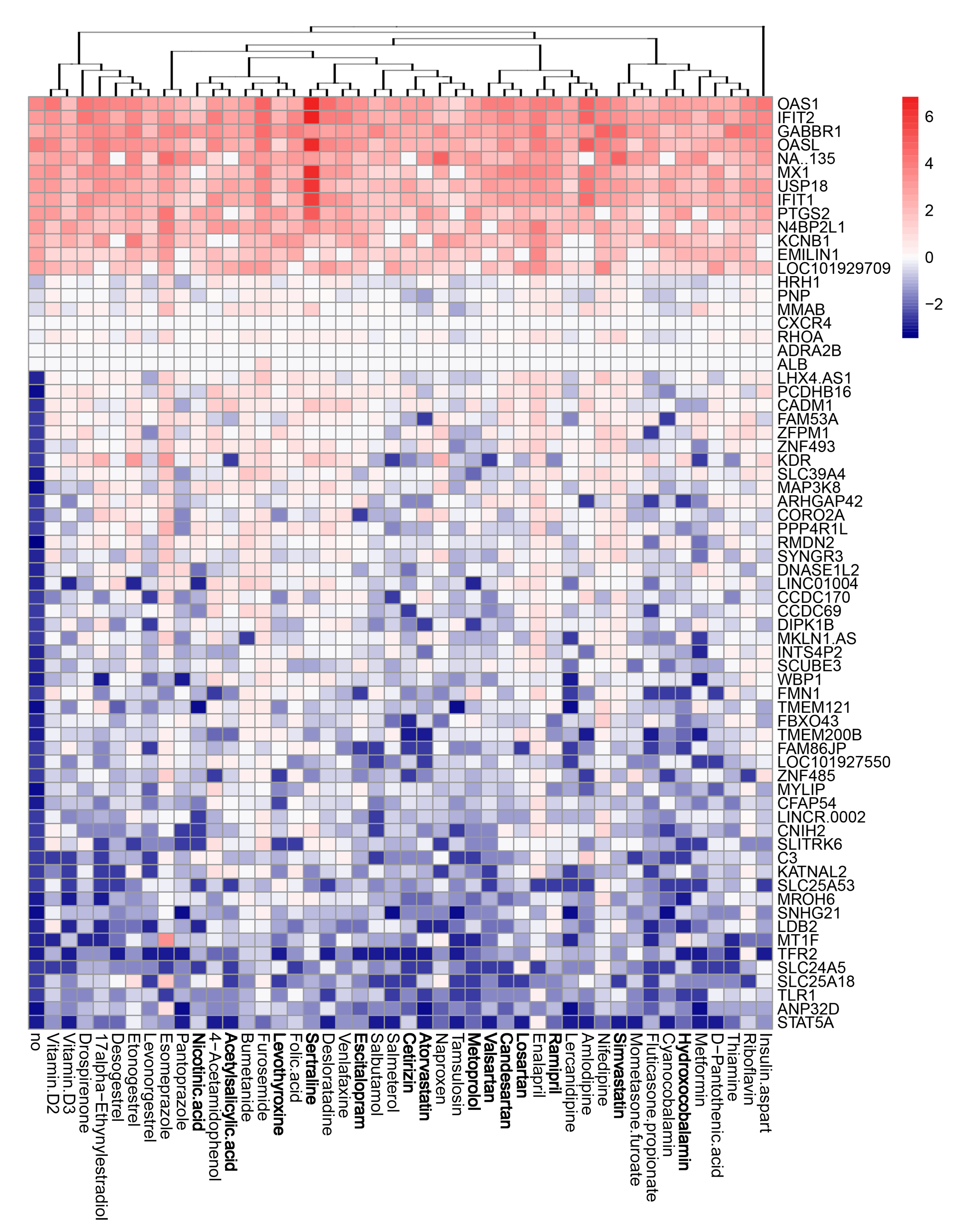
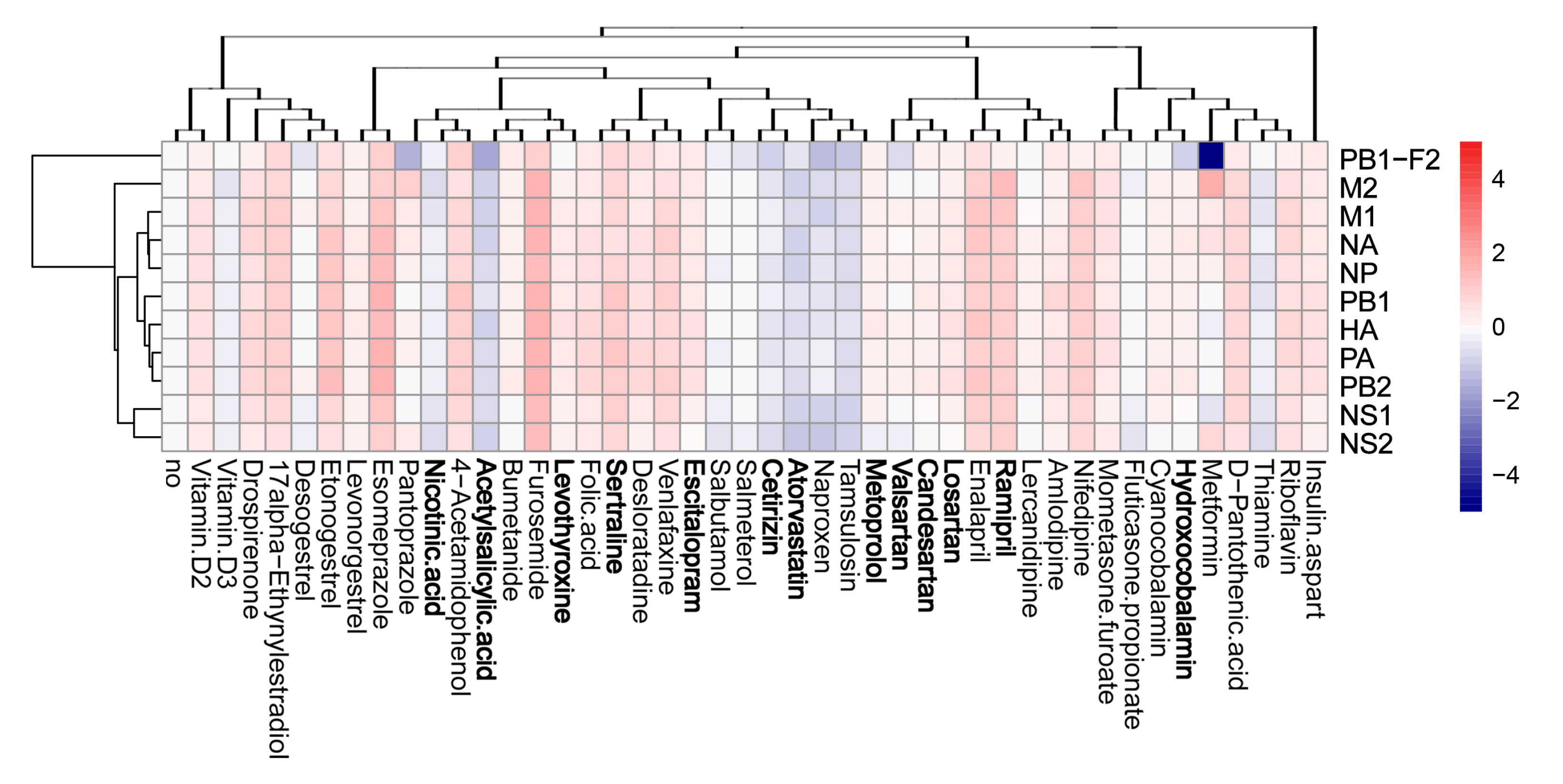
3.4. Active Components of Commonly Prescribed Medicines Affect Gene Expression in Mock- and FLUAV-Infected RPE Cells
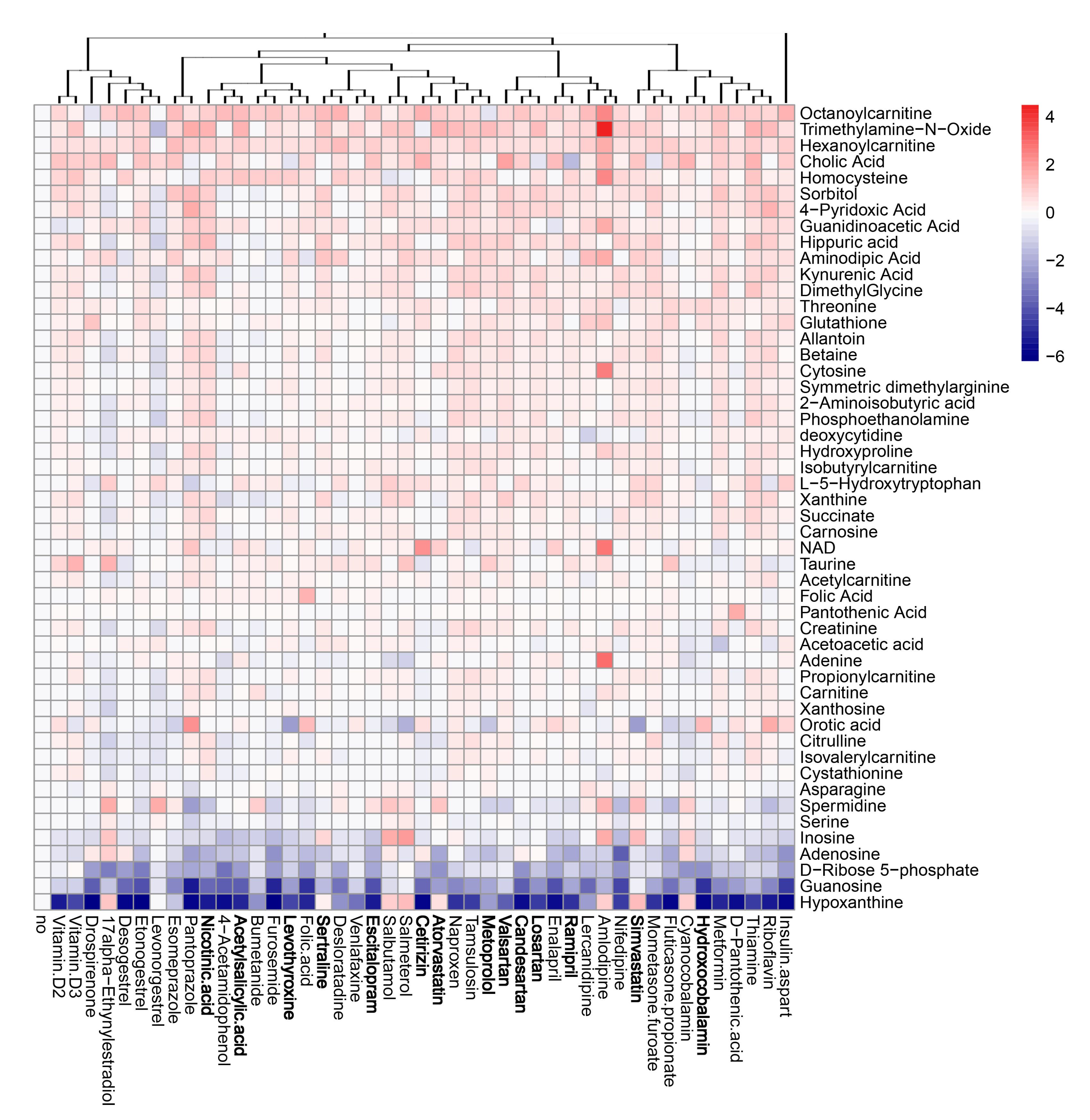
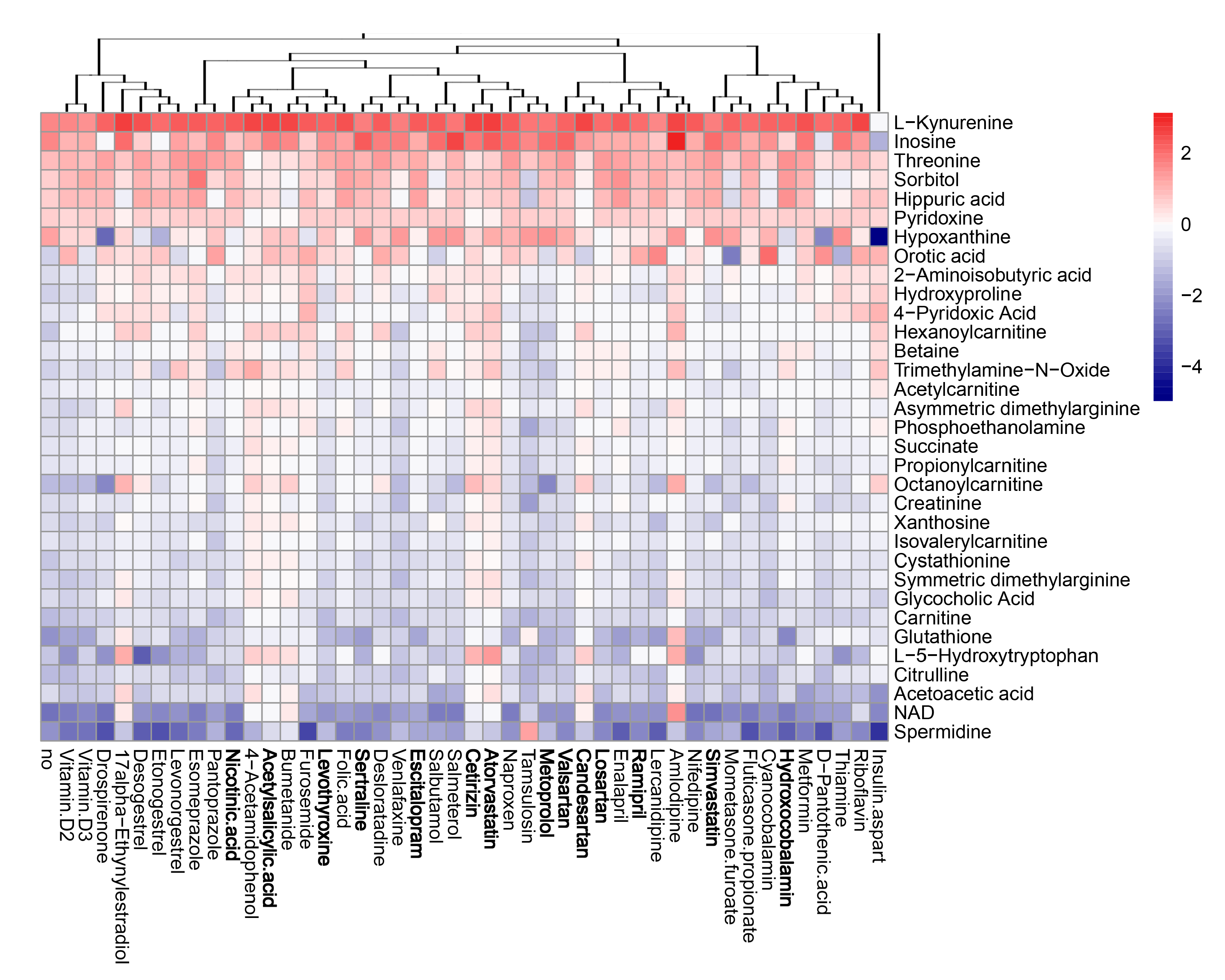
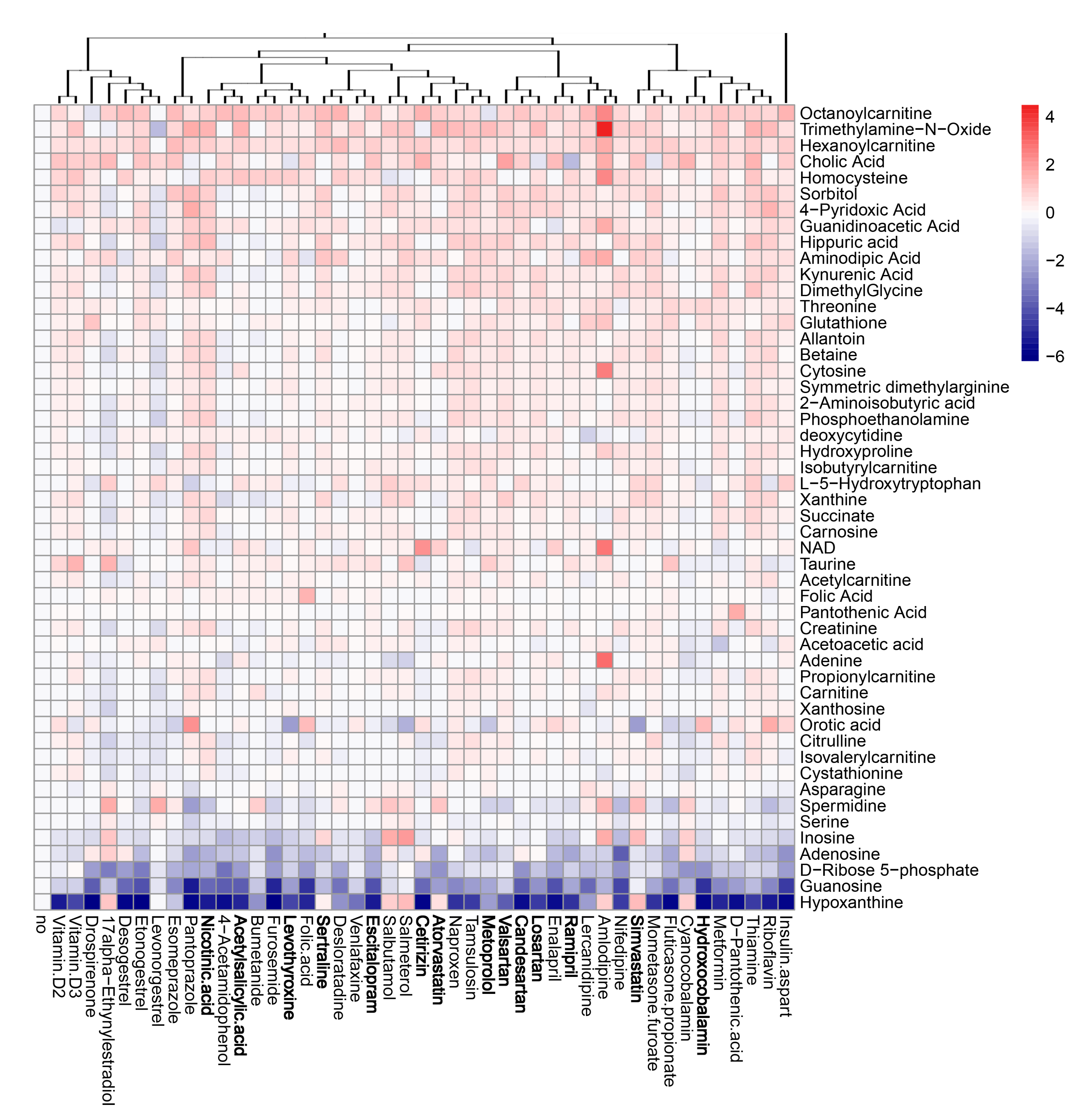
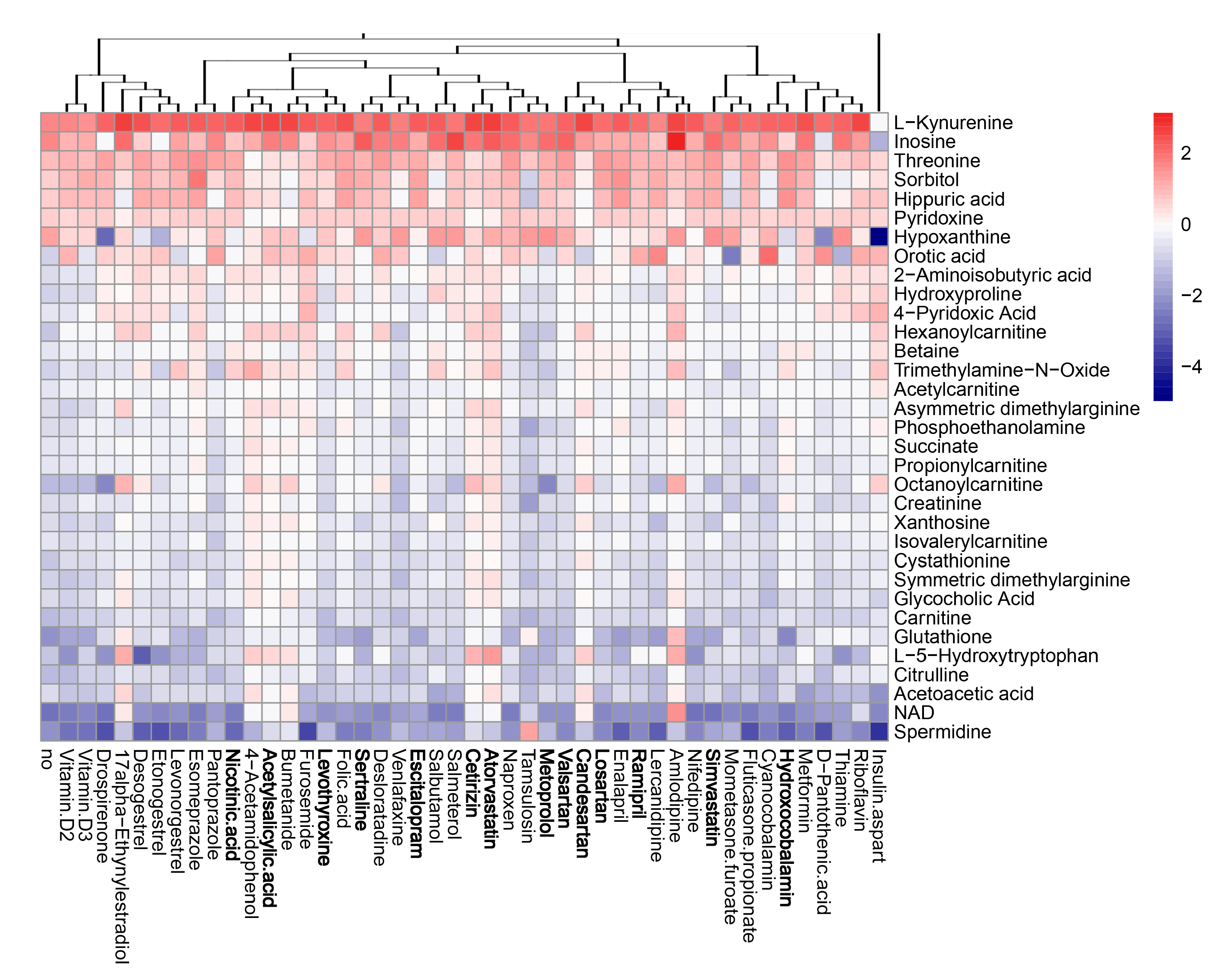
3.5. Active Components of Commonly Prescribed Medicines Affect Metabolism of Mock- and FLUAV-Infected RPE Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, A.; Madhavan, M.V.; Poterucha, T.J.; DeFilippis, E.M.; Hennessey, J.A.; Redfors, B.; Eckhardt, C.; Bikdeli, B.; Platt, J.; Nalbandian, A.; et al. Association between antecedent statin use and decreased mortality in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 1325. [Google Scholar] [CrossRef] [PubMed]
- Mauskopf, J.; Klesse, M.; Lee, S.; Herrera-Taracena, G. The burden of influenza complications in different high-risk groups: A targeted literature review. J. Med. Econ. 2013, 16, 264–277. [Google Scholar] [CrossRef]
- Asai, N.; Ohkuni, Y.; Komatsu, A.; Matsunuma, R.; Nakashima, K.; Ando, K.; Iwasaki, T.; Yasui, D.; Misawa, M.; Otsuka, Y.; et al. A case of asthma-complicated influenza myocarditis. J. Infect. Chemother. 2011, 17, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.Y.; Kainov, D.E. Influenza virus infection, interferon response, viral counter-response, and apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef] [Green Version]
- Anastasina, M.; Le May, N.; Bugai, A.; Fu, Y.; Soderholm, S.; Gaelings, L.; Ohman, T.; Tynell, J.; Kyttanen, S.; Barboric, M.; et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim. Biophys. Acta. 2016, 1859, 1440–1448. [Google Scholar] [CrossRef] [Green Version]
- Anastasina, M.; Schepens, B.; Soderholm, S.; Nyman, T.A.; Matikainen, S.; Saksela, K.; Saelens, X.; Kainov, D.E. The C terminus of NS1 protein of influenza A/WSN/1933(H1N1) virus modulates antiviral responses in infected human macrophages and mice. J. Gen. Virol 2015, 96, 2086–2091. [Google Scholar] [CrossRef]
- Kainov, D.E.; Muller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential effects of NS1 proteins of human pandemic H1N1/2009, avian highly pathogenic H5N1, and low pathogenic H5N2 influenza A viruses on cellular pre-mRNA polyadenylation and mRNA translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Kulesskiy, E.; Krpina, K.; Lou, G.; Aman, Y.; Bugai, A.; Aasumets, K.; Akimov, Y.; Bulanova, D.; Gildemann, K.; et al. Chemical, physical and biological triggers of evolutionary conserved Bcl-xL-mediated apoptosis. Cancers 2020, 12, 1694. [Google Scholar] [CrossRef]
- Kakkola, L.; Denisova, O.V.; Tynell, J.; Viiliainen, J.; Ysenbaert, T.; Matos, R.C.; Nagaraj, A.; Ohman, T.; Kuivanen, S.; Paavilainen, H.; et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013, 4, e742. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, S.; Fu, Y.; Gaelings, L.; Belanov, S.; Yetukuri, L.; Berlinkov, M.; Cheltsov, A.V.; Anders, S.; Aittokallio, T.; Nyman, T.A.; et al. Multi-omics studies towards novel modulators of influenza A virus-host interaction. Viruses 2016, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, K.H.; Kakkola, L.; Nagaraj, A.S.; Cheltsov, A.V.; Anastasina, M.; Kainov, D.E. Emerging cellular targets for influenza antiviral agents. Trends Pharmacol. Sci. 2012, 33, 89–99. [Google Scholar] [CrossRef]
- Muller, K.H.; Kainov, D.E.; El Bakkouri, K.; Saelens, X.; De Brabander, J.K.; Kittel, C.; Samm, E.; Muller, C.P. The proton translocation domain of cellular vacuolar ATPase provides a target for the treatment of influenza A virus infections. Br. J. Pharmacol. 2011, 164, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, S.; Kainov, D.E.; Ohman, T.; Denisova, O.V.; Schepens, B.; Kulesskiy, E.; Imanishi, S.Y.; Corthals, G.; Hintsanen, P.; Aittokallio, T.; et al. Phosphoproteomics to characterize host response during influenza A virus infection of human macrophages. Mol. Cell Proteom. 2016, 15, 3203–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisova, O.V.; Soderholm, S.; Virtanen, S.; Von Schantz, C.; Bychkov, D.; Vashchinkina, E.; Desloovere, J.; Tynell, J.; Ikonen, N.; Theisen, L.L.; et al. Akt inhibitor MK2206 prevents influenza pH1N1 virus infection in vitro. Antimicrob. Agents Chemother. 2014, 58, 3689–3696. [Google Scholar] [CrossRef] [Green Version]
- Bajimaya, S.; Frankl, T.; Hayashi, T.; Takimoto, T. Cholesterol is required for stability and infectivity of influenza A and respiratory syncytial viruses. Virology 2017, 510, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Denisova, O.V.; Kakkola, L.; Feng, L.; Stenman, J.; Nagaraj, A.; Lampe, J.; Yadav, B.; Aittokallio, T.; Kaukinen, P.; Ahola, T.; et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza A virus infection. J. Biol. Chem. 2012, 287, 35324–35332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marakasova, E.S.; Eisenhaber, B.; Maurer-Stroh, S.; Eisenhaber, F.; Baranova, A. Prenylation of viral proteins by enzymes of the host: Virus-driven rationale for therapy with statins and FT/GGT1 inhibitors. Bioessays 2017, 39, 1700014. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, G.; Ye, X. Cyclin T1/CDK9 interacts with influenza A virus polymerase and facilitates its association with cellular RNA polymerase II. J. Virol. 2010, 84, 12619–12627. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, E.; Webster, R.G. Unidirectional RNA polymerase I-polymerase II transcription system for the generation of influenza A virus from eight plasmids. J. Gen. Virol. 2000, 81, 2843–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaelings, L.; Soderholm, S.; Bugai, A.; Fu, Y.; Nandania, J.; Schepens, B.; Lorey, M.B.; Tynell, J.; Vande Ginste, L.; Le Goffic, R.; et al. Regulation of kynurenine biosynthesis during influenza virus infection. FEBS J. 2017, 284, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Namdari, H.; Arjeini, Y.; Mousavi, M.J.; Rezaei, F. Metabolic host response and therapeutic approaches to influenza infection. Cell Mol. Biol. Lett. 2020, 25, 15. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, K.; Min, J.; Chen, C.; Cao, Y.; Ding, C.; Liu, W.; Li, J. Metabolomic analysis of influenza A Virus A/WSN/1933 (H1N1) infected A549 cells during first cycle of viral replication. Viruses 2019, 11, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zusinaite, E.; Ianevski, A.; Niukkanen, D.; Poranen, M.M.; Bjoras, M.; Afset, J.E.; Tenson, T.; Velagapudi, V.; Merits, A.; Kainov, D.E. A systems approach to study immuno-and neuro-modulatory properties of antiviral agents. Viruses 2018, 10, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ianevski, A.; Yao, R.; Zusinaite, E.; Lysvand, H.; Oksenych, V.; Tenson, T.; Bjørås, M.; Kainov, D. Active Components of Commonly Prescribed Medicines Affect Influenza A Virus–Host Cell Interaction: A Pilot Study. Viruses 2021, 13, 1537. https://doi.org/10.3390/v13081537
Ianevski A, Yao R, Zusinaite E, Lysvand H, Oksenych V, Tenson T, Bjørås M, Kainov D. Active Components of Commonly Prescribed Medicines Affect Influenza A Virus–Host Cell Interaction: A Pilot Study. Viruses. 2021; 13(8):1537. https://doi.org/10.3390/v13081537
Chicago/Turabian StyleIanevski, Aleksandr, Rouan Yao, Eva Zusinaite, Hilde Lysvand, Valentyn Oksenych, Tanel Tenson, Magnar Bjørås, and Denis Kainov. 2021. "Active Components of Commonly Prescribed Medicines Affect Influenza A Virus–Host Cell Interaction: A Pilot Study" Viruses 13, no. 8: 1537. https://doi.org/10.3390/v13081537
APA StyleIanevski, A., Yao, R., Zusinaite, E., Lysvand, H., Oksenych, V., Tenson, T., Bjørås, M., & Kainov, D. (2021). Active Components of Commonly Prescribed Medicines Affect Influenza A Virus–Host Cell Interaction: A Pilot Study. Viruses, 13(8), 1537. https://doi.org/10.3390/v13081537