
Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Cloning
2.3. Site-Directed Mutagenesis
2.4. Lentivirus Production and Transduction
2.5. Transfections
2.6. Western Blot
2.7. Isolation of RNA, cDNA Synthesis and RTqPCR
2.8. Luciferase Assays
2.9. ChIP Assays
2.10. In Silico Sequence Analysis
2.11. Statistical Analysis
3. Results
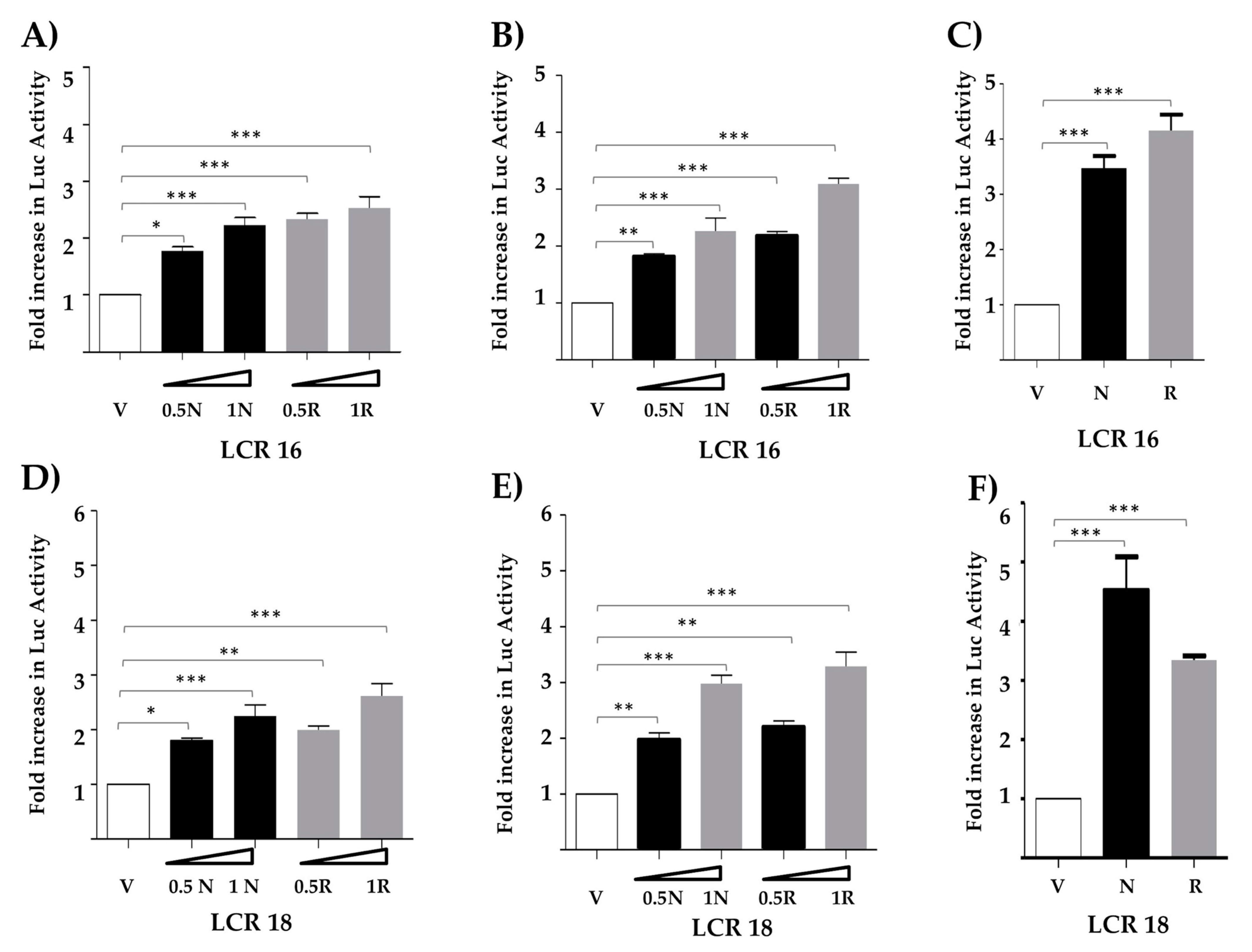
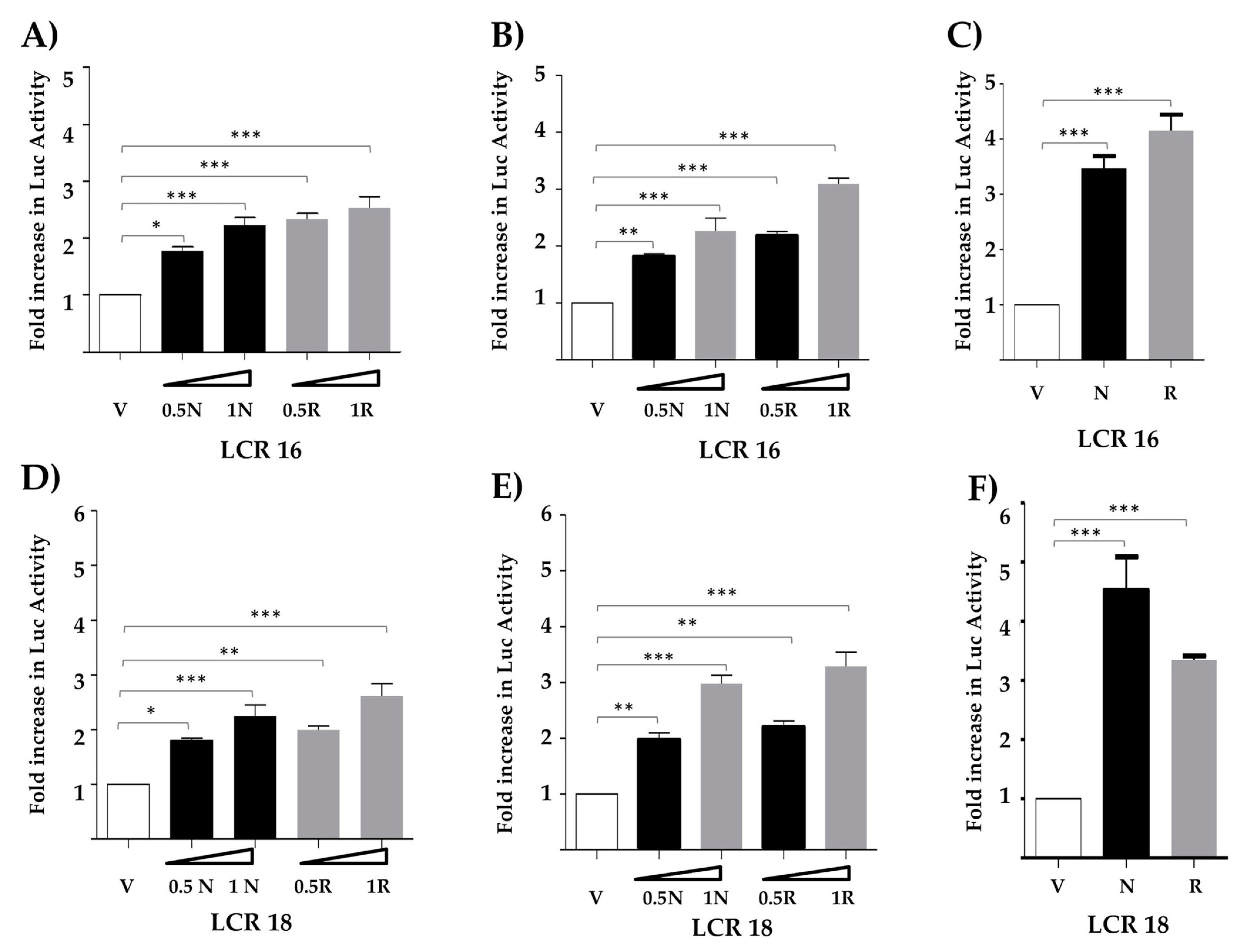
3.1. Nanog Increases Transcriptional Activity from the LCR of HPV Types 16 and 18
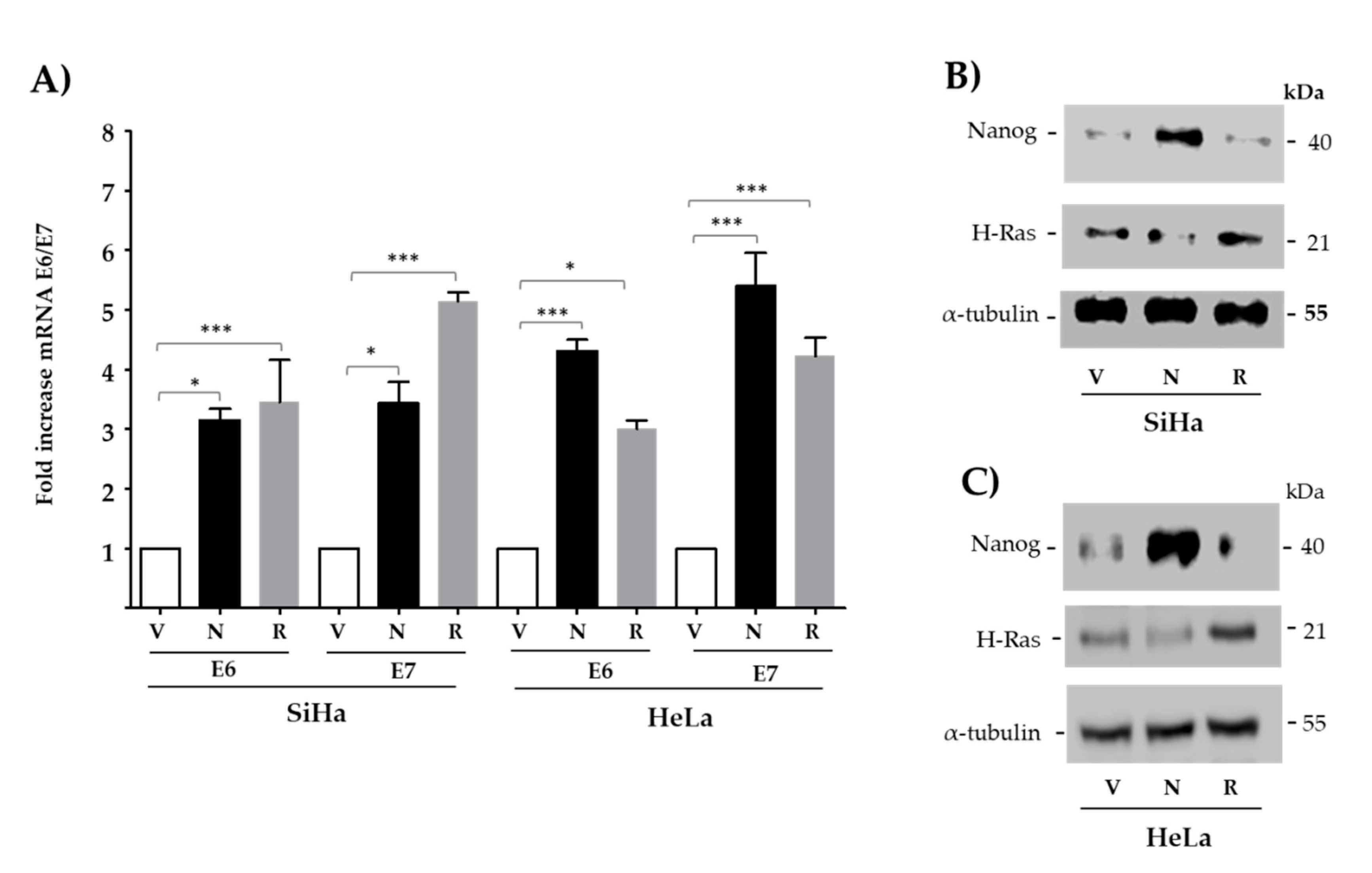
3.2. The Expression of E6 and E7 Is Increased by Exogenous Nanog in Both HPV16/18-Positive Cancer-Derived Cell Lines
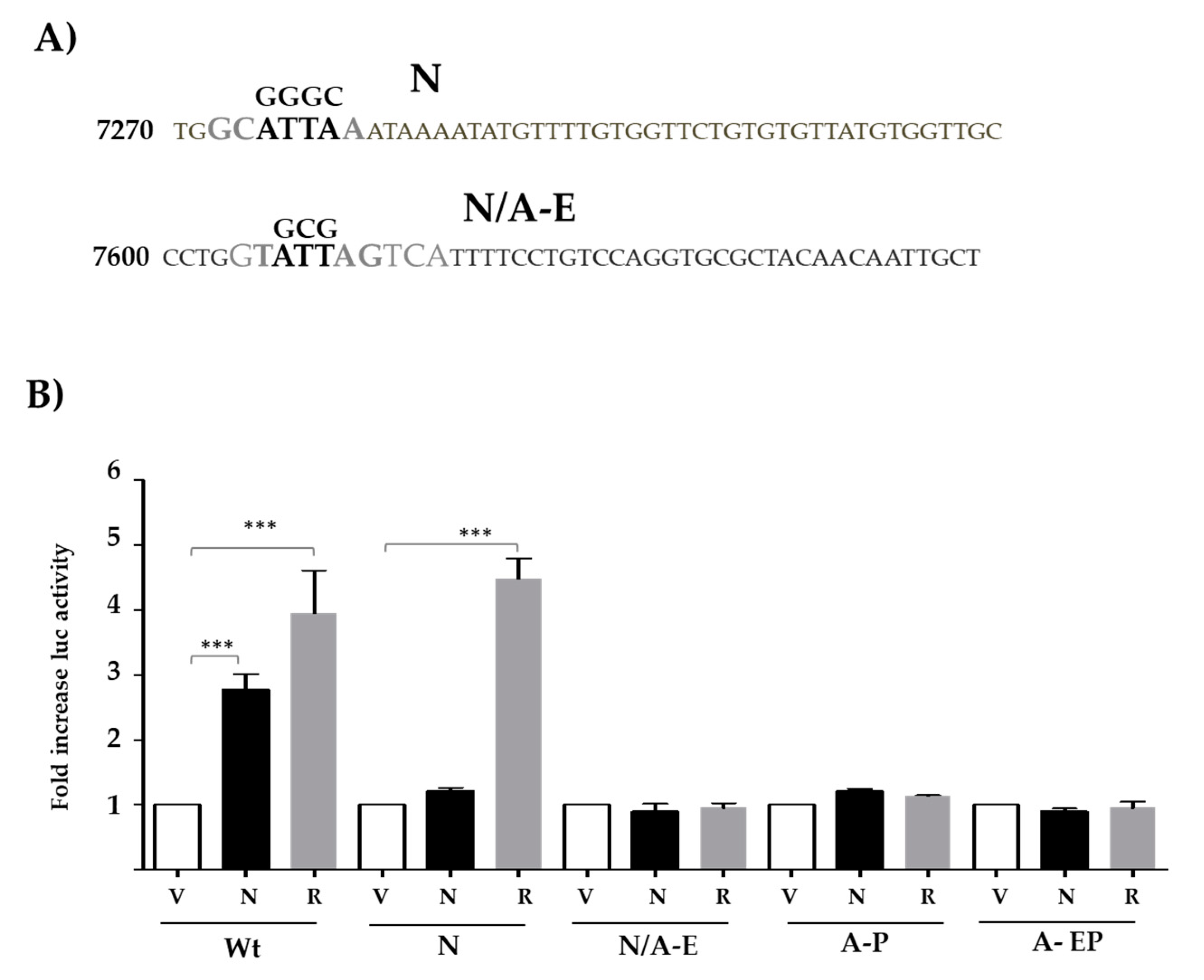
3.3. Nanog- and AP1-Binding Sites Are Necessary for LCR-Mediated Activation Along
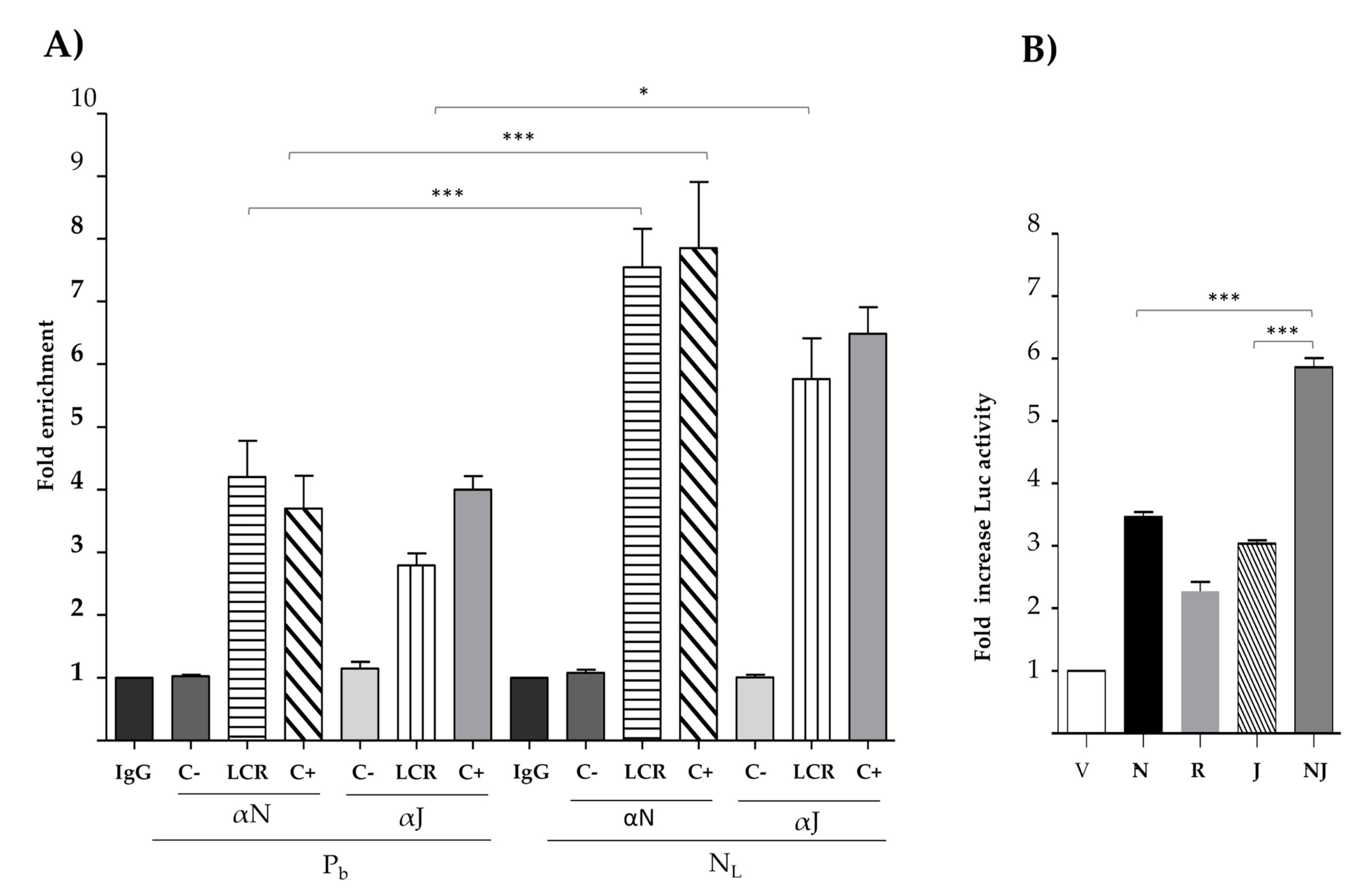
3.4. Nanog Binds In Vivo to the HPV18 LCR and Cooperates with C-Jun to Increase Transcriptional Activity
3.5. Identification of Nanog-Binding Sites in LCRs of Different Alpha- and Betapapillomaviruses Associated with Cancer Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Bosch, F.X.; Burchell, A.N.; Schiffman, M.; Giuliano, A.R.; de Sanjose, S.; Bruni, L.; Tortolero-Luna, G.; Kjaer, S.K.; Munoz, N. Epidemiology and natural history of human papillomavirus infections and type-specific implications in cervical neoplasia. Vaccine 2008, 26, K1–K16. [Google Scholar] [CrossRef] [PubMed]
- Burley, M.; Roberts, S.; Parish, J.L. Epigenetic Regulation of Human Papillomavirus Transcription in the Productive Virus Life Cycle. Semin. Immunopathol. 2020, 42, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, M.; Bernard, H.U. Oct-1 Activates the Epithelial-Specific Enhancer of Human Papillomavirus Type 16 via a Synergistic Interaction with NFI at a Conserved Composite Regulatory Element. Virology 1995, 207, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prati, B.; Marangoni, B.; Boccardo, E. Human Papillomavirus and Genome Instability: From Productive Infection to Cancer. Clinics 2018, 73, e539s. [Google Scholar] [CrossRef]
- Chesters, P.M.; McCance, D.J. Human papillomavirus types 6 and 16 in cooperation with Ha-ras transform secondary rat embryo fibroblasts. J. Gen. Virol. 1989, 70, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Chong, T.; Chan, W.K.; Bernard, H.U. Transcriptional activation of human papillomavirus 16 by nuclear factor I, AP1, steroid receptors and a possibly novel transcription factor, PVF: A model for the composition of genital papillomavirus enhancers. Nucleic Acids Res. 1990, 18, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, E.E.; Cowan, C.E.; Chatterjee, I.; Malik, A.B.; Wary, K.K. NANOG induction of fetal liver kinase-1 (FLK1) transcription regulates endothelial cell proliferation and angiogenesis. Blood 2011, 117, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Li, Y.; Cao, X.; Du, J.; Huang, X. NANOG regulates epithelial-mesenchymal transition and chemoresistance in ovarian cancer. Biosci. Rep. 2017, 37, BSR20160247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.H.; Choi, C.H.; Lee, H.J.; Oh, S.J.; Woo, S.R.; Hong, S.O.; Noh, K.H.; Cho, H.; Chung, E.J.; Kim, J.H.; et al. Upregulation by NANOG Promotes Multidrug Resistance and a Stem-like Phenotype in Immune Edited Tumor Cells. Cancer Res. 2017, 77, 5039–5053. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, J.; Chen, S.; Fang, C.; Zhang, X.; Luo, Z. Prognostic significance of NANOG expression in solid tumors: A meta-analysis. OncoTargets Ther. 2018, 11, 5515–5526. [Google Scholar] [CrossRef] [Green Version]
- Dehghan Harati, M.; Rodemann, H.P.; Toulany, M. Nanog Signaling Mediates Radioresistance in ALDH-Positive Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Cho, H.; Hong, S.O.; Oh, S.J.; Lee, H.J.; Cho, E.; Woo, S.R.; Song, J.S.; Chung, J.Y.; Son, S.W.; et al. LC3B upregulation by NANOG promotes immune resistance and stem-like property through hyperactivation of EGFR signaling in immune-refractory tumor cells. Autophagy 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Basati, G.; Mohammadpour, H.; Emami Razavi, A. Association of High Expression Levels of SOX2, NANOG, and OCT4 in Gastric Cancer Tumor Tissues with Progression and Poor Prognosis. J. Gastrointest. Cancer 2020, 51, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Nath, N.; Mishra, P.; Jha, A.; Nagini, S.; Mishra, R. Pluripotency Transcription Factor Nanog and Its Association with Overall Oral Squamous Cell Carcinoma Progression, Cisplatin-Resistance, Invasion and Stemness Acquisition. Head Neck 2020, 42, 3282–3294. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Park, M.J.; Choi, S.I.; In, K.H.; Kim, C.H.; Lee, S.H. NANOG as an adverse predictive marker in advanced non–small cell lung cancer treated with platinum-based chemotherapy. OncoTargets Ther. 2017, 10, 4625–4633. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Kang, Y.H.; Byun, J.H.; Jang, S.J.; Rho, G.J.; Lee, J.S.; Park, B.W. Midkine and NANOG Have Similar Immunohistochemical Expression Patterns and Contribute Equally to an Adverse Prognosis of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, J.P.; Villaronga, M.A.; Menéndez, S.T.; Hermida-Prado, F.; Quer, M.; Vilaseca, I.; Allonca, E.; Mallo, D.P.; Astudillo, A.; García-Pedrero, J.M. A Novel Role for Nanog As An Early Cancer Risk Marker in Patients with Laryngeal Precancerous Lesions. Sci. Rep. 2017, 7, 11110. [Google Scholar] [CrossRef] [PubMed]
- Kenda Šuster, N.; Frković Grazio, S.; Virant-Klun, I.; Verdenik, I.; Smrkolj, Š. Cancer Stem Cell-Related Marker NANOG Expression in Ovarian Serous Tumors: A Clinicopathological Study of 159 Cases. Int. J. Gynecol. Cancer 2017, 27, 2006–2013. [Google Scholar] [CrossRef] [PubMed]
- Md Akhir, M.K.A.; Hussin, H.; Veerakumarasivam, A.; Choy, C.S.; Abdullah, M.A.; Abd Ghani, F. Immunohistochemical expression of NANOG in urothelial carcinoma of the bladder. Malays. J. Pathol. 2017, 39, 227–234. [Google Scholar] [PubMed]
- Soni, P.; Qayoom, S.; Husain, N.; Kumar, P.; Chandra, A.; Ojha, B.K.; Gupta, R.K. CD24 and Nanog expression in Stem Cells in Glioblastoma: Correlation with Response to Chemoradiation and Overall Survival. Asian Pac. J. Cancer Prev. 2017, 18, 2215–2219. [Google Scholar] [CrossRef]
- Kuciak, M.; Mas, C.; Borges, I.; Sánchez-Gómez, P.; Ruiz, A. Chimeric NANOG repressors inhibit glioblastoma growth in vivo in a context-dependent manner. Sci. Rep. 2019, 9, 3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Zhao, T.; Ge, H.; Xu, Y.; Ren, S.; Yue, C.; Li, G.; Wu, J. The clinicopathological and prognostic value of Nanog in human gastrointestinal luminal cancer: A meta-analysis. Int. J. Surg. 2018, 53, 193–200. [Google Scholar] [CrossRef]
- Wang, H.; Liu, B.; Wang, J.; Li, J.; Gong, Y.; Li, S.; Wang, C.; Cui, B.; Xue, X.; Yang, M.; et al. Reduction of NANOG Mediates the Inhibitory Effect of Aspirin on Tumor Growth and Stemness in Colorectal Cancer. Cell Physiol. Biochem. 2017, 441051–441063. [Google Scholar] [CrossRef]
- Emadian Saravi, O.; Naghshvar, F.; Torabizadeh, Z.; Sheidaei, S. Immunohistochemical Expression of Nanog and Its Relation with Clinicopathologic Characteristics in Breast Ductal Carcinoma. Iran. Biomed. J. 2019, 23, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Zhou, C.; Cheng, Q.; Shen, J.; Chen, H. Stem-cell-abundant proteins Nanog, Nucleostemin and Musashi1 are highly expressed in malignant cervical epithelial cells. BMC Cancer 2008, 8, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, T.-T.; Liu, S.-Y.; Zheng, P.-S. Cytoplasmic NANOG-positive stromal cells promote human cervical cancer progression. Am. J. Pathol. 2012, 181, 652–661. [Google Scholar] [CrossRef]
- Liu, S.; Minaguchi, T.; Lachkar, B.; Zhang, S.; Xu, C.; Tenjimbayashi, Y.; Shikama, A.; Tasaka, N.; Akiyama, A.; Sakurai, M.; et al. Separate analysis of human papillomavirus E6 and E7 messenger RNAs to predict cervical neoplasia progression. PLoS ONE 2018, 13, e0193061. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Deodhar, K.; Pai, V.; Pant, S.; Rathod, N.; Goda, J.S.; Sudhalkar, N.; Pandey, P.; Waghmare, S.; Engineer, R.; et al. CD44, and Outcomes Following Chemoradiation in Locally Advanced Cervical Cancer: Results From a Prospective Study. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Organista-Nava, J.; Gómez-Gómez, Y.; Ocadiz-Delgado, R.; García-Villa, E.; Bonilla-Delgado, J.; Lagunas-Martínez, A.; Tapia, J.S.; Lambert, P.F.; García-Carrancá, A.; Gariglio, P. The HPV16 E7 oncoprotein increases the expression of Oct3/4 and stemness-related genes and augments cell self-renewal. Virology 2016, 499, 230–242. [Google Scholar] [CrossRef] [PubMed]
- García-Carrancá, A.; Thierry, F.; Yaniv, M. Interplay of viral and cellular proteins along the long control region of human papillomavirus type 18. J. Virol. 1988, 62, 4321–4330. [Google Scholar] [CrossRef] [Green Version]
- Medina-Martínez, O.; Vallejo, V.; Guido, M.C.; García-Carrancá, A. Ha-ras oncogene–induced transcription of human papillomavirus type 18 E6 and E7 oncogenes. Mol. Carcinog. 1997, 19, 83–90. [Google Scholar] [CrossRef]
- Priyadarshini, R.; Hussain, M.; Attri, P.; Kaur, E.; Tripathi, V.; Priya, S.; Dhapola, P.; Saha, D.; Madhavan, V.; Chowdhury, S.; et al. BLM Potentiates c-Jun Degradation and Alters Its Function as an Oncogenic Transcription Factor. Cell Rep. 2018, 24, 947–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, J.P.; Carrillo-Beltrán, D.; Aedo-Aguilera, V.; Calaf, G.M.; León, O.; Maldonado, E.; Tapia, J.C.; Boccardo, E.; Ozbun, M.A.; Aguayo, F. Tobacco Exposure Enhances Human Papillomavirus 16 Oncogene Expression via EGFR/PI3K/Akt/c-Jun Signaling Pathway in Cervical Cancer Cells. Front Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Scarth, J.A.; Patterson, M.R.; Christopher, W.; Georgia, W.C.; Barba-Moreno, H.D.; Macdonald, A. E6-mediated activation of JNK drives EGFR signalling to promote proliferation and viral oncoprotein expression in cervical cancer. Cell Death Differ. 2020, 28, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.I.; Santana, C.; Rojas, E.; Hernández, S.; Ostrosky-Wegman, P.; García-Carrancá, A. Induced mitotic death of HeLa cells by abnormal expression of c-H-ras. Mutat. Res. Mol. Mech. Mutagen. 1997, 349, 173–182. [Google Scholar] [CrossRef]
- Martínez-Ramírez, I.; Del-Castillo-Falconi, V.; Mitre-Aguilar, I.B.; Amador-Molina, A.; Carrillo-García, A.; Langley, E.; Zentella-Dehesa, A.; Soto-Reyes, E.; García-Carrancá, A.; Herrera, L.A.; et al. SOX2 as a New Regulator of HPV16 Transcription. Viruses 2017, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F.; Spyrou, G.; Yaniv, M.; Howley, P. Two AP1 Sites Binding JunB Are Essential for Human Papillomavirus Type 18 Transcription in Keratinocytes. J. Virol. 1992, 66, 3740–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, S.I.; Bourachot, B.; Yaniv, M. Both Jun and Fos contribute to transcription activation by the heterodimer. Oncogene 1990, 5, 39–46. [Google Scholar] [PubMed]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xiong, F.; Zhou, Y.; Wu, X.; Liu, F.; Xue, S.; Chen, H. NANOG upregulates c-Jun oncogene expression through binding the c-Jun promoter. Mol. Carcinog. 2015, 54, 1407–1416. [Google Scholar] [CrossRef]
- Das, S.; Jena, S.; Kim, E.M.; Zavazava, N.; Levasseur, D.N. Transcriptional Regulation of Human NANOG by Alternate Promoters in Embryonic Stem Cells. J. Stem Cell Res. Ther. 2012, 10, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, G.; Valencia-González, H.A.; Pérez-Montiel, D.; Muñoz, F.; Ocadiz-Delgado, R.; Fernández-Retana, J.; Pérez-Plasencia, C.; Reséndis-Antonio, O.; Gariglio, P.; García-Carrancá, A. Genes Involved in the Transcriptional Regulation of Pluripotency Are Expressed in Malignant Tumors of the Uterine Cervix and Can Induce Tumorigenic Capacity in a Nontumorigenic Cell Line. Stem Cells Int. 2019, 7683817. [Google Scholar] [CrossRef]
- Han, G.H.; Cho, H. 203 High expression of Nanog and CRY1 is involved with tumor progression and poor prognosis in patients with cervical cancer. Int. J. Gynecol. Cancer 2020, 30, A7. [Google Scholar] [CrossRef]
- Gunasekharan, V.K.; Li, Y.; Andrade, J.; Laimins, L.A. Post-Transcriptional Regulation of KLF4 by High-Risk Human Papillomaviruses Is Necessary for the Differentiation-Dependent Viral Life Cycle. PLoS Pathog. 2016, 12, e1005747. [Google Scholar] [CrossRef] [PubMed]
- Sichero, L.; Sobrinho, J.S.; Villa, L.L. Identification of Novel Cellular Transcription Factors that Regulate Early Promoters of Human Papillomavirus Types 18 and 16. J. Infect. Dis. 2012, 206, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Sánchez, E.; Santiago-López, L.; Cruz-Domínguez, V.B.; Toledo-Guzmán, M.E. Hernández-Cueto, D.; Muñiz-Hernández, S.; Garrido, E.; Cantú De León, D.; García-Carrancá. A. Characterization of cervical cancer stem cell-like cells: Phenotyping, stemness, and human papilloma virus co-receptor expression. OncoTarget 2016, 7, 31943–31954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, E.E.; Babaei-Jadidi, R.; Saadeddin, A.; Spencer-Dene, B.; Hossaini, S.; Abuzinadah, M.; Li, N.; Fadhil, W.; Ilyas, M.; Bonnet, D.; et al. Embryonic NANOG Activity Defines Colorectal Cancer Stem Cells and Modulates through AP1- and TCF-Dependent Mechanisms. Stem Cells 2012, 30, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Garces de los Fayos Alonso, I.; Liang, H.C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, J.Z.; Yuan, X.H.; Adler-Storthz, K.; Che, Z. An AP-1 Binding Site Mutation in HPV-16 LCR Enhances E6/E7 Promoter Activity in Human Oral Epithelial Cells. Virus Genes 2002, 24, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Klumpp, D.J.; Inoue, M.; Kanaya, T.; Laimins, L.A. Expression of AP1 during cellular differentiation determines human papillomavirus E6/E7 expression in stratified epithelial cells. J. Gen. Virol. 1997, 78, 401–411. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | Type | Position | Nanog-Putative- Binding Sites | Bases To +1 |
|---|---|---|---|---|---|
| Alpha | 9 | 16 | 7171–7177 | 5′- tatgttGAATTAGtgttgt-3´ | 823 |
| 31 | 7813–7819 | 5′- tgtcatGCATTATaaataa-3´ | 199 | ||
| 7861–7867 | 5′- atatagGTATTACaccgtt-3´ | 152 | |||
| 35 | 7565–7771 | 5′- cttgcaGTATTAGtcattt-3´ | 386 | ||
| 7761–7767 | 5′- ctcactGTATTACacattg-3´ | 193 | |||
| 52 | 7334–7340 | 5′- gtgtatGTATTAAtaaagta-3´ | 705 | ||
| 5 | 51 | 7241–7247 | 5′- atgtggGTATTACattatc-3´ | 703 | |
| 6 | 56 | 7617–7623 | 5′- caccctGTATTACtcacag-3´ | 322 | |
| 7 | 18 | 7272–7278 | 5′- tatgtgGCATTAAataaaa-3´ | 683 | |
| 7605–7611 | 5′- cacctgGTATTAGtcattt-3´ | 350 | |||
| 45 | 7603–7609 | 5′- cacctgGTATTAGtcattt-3´ | 350 | ||
| 12 | MmPV2 | 7206–7212 | 5′- gttaaaGTATTAAtaaagg-3´ | 664 | |
| Beta | 1 | 5 | 7669–7675 | 5′- atgctcGGATTAGggacct-3´ | 268 |
| 8 | 7576–7582 | 5′- atgctcGGATTAGgtcgcc-3´ | 267 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Tejeda, Y.; Guido-Jiménez, M.C.; López-Carbajal, H.; Amador-Molina, A.; Méndez-Martínez, R.; Gariglio-Vidal, P.; Lizano, M.; García-Carrancá, A. Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18. Viruses 2021, 13, 1482. https://doi.org/10.3390/v13081482
Díaz-Tejeda Y, Guido-Jiménez MC, López-Carbajal H, Amador-Molina A, Méndez-Martínez R, Gariglio-Vidal P, Lizano M, García-Carrancá A. Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18. Viruses. 2021; 13(8):1482. https://doi.org/10.3390/v13081482
Chicago/Turabian StyleDíaz-Tejeda, Yakelin, Miriam C. Guido-Jiménez, Helga López-Carbajal, Alfredo Amador-Molina, Rocío Méndez-Martínez, Patricio Gariglio-Vidal, Marcela Lizano, and Alejandro García-Carrancá. 2021. "Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18" Viruses 13, no. 8: 1482. https://doi.org/10.3390/v13081482
APA StyleDíaz-Tejeda, Y., Guido-Jiménez, M. C., López-Carbajal, H., Amador-Molina, A., Méndez-Martínez, R., Gariglio-Vidal, P., Lizano, M., & García-Carrancá, A. (2021). Nanog, in Cooperation with AP1, Increases the Expression of E6/E7 Oncogenes from HPV Types 16/18. Viruses, 13(8), 1482. https://doi.org/10.3390/v13081482