
Targeting Viral Surface Proteins through Structure-Based Design


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
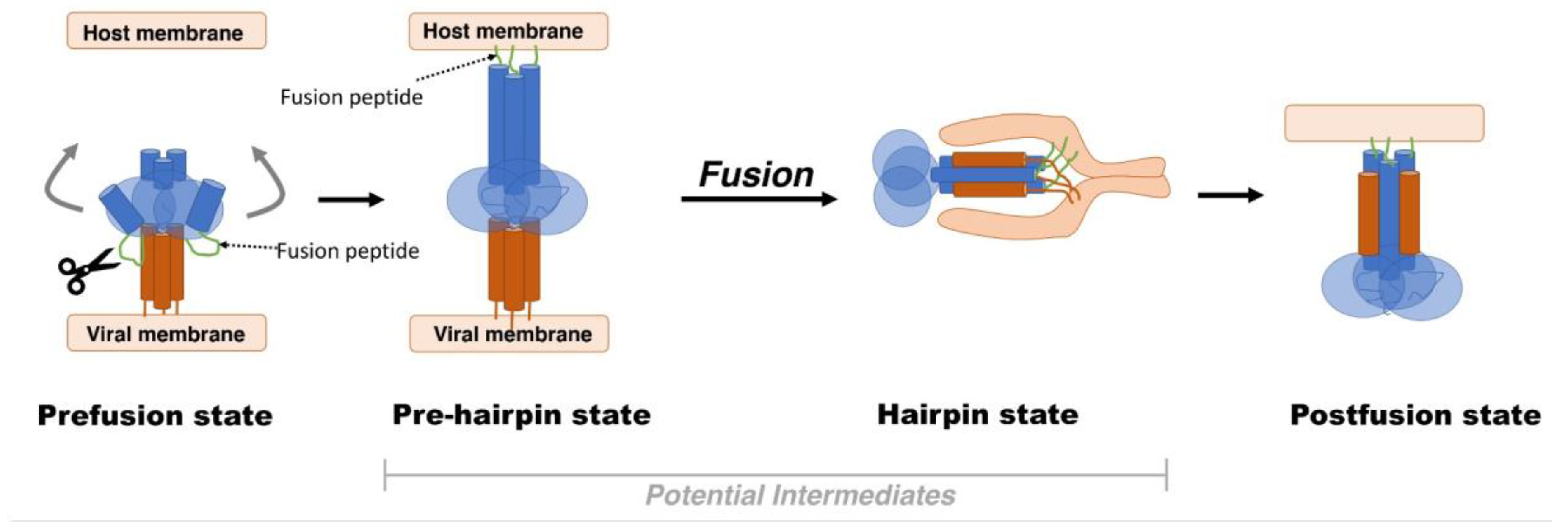
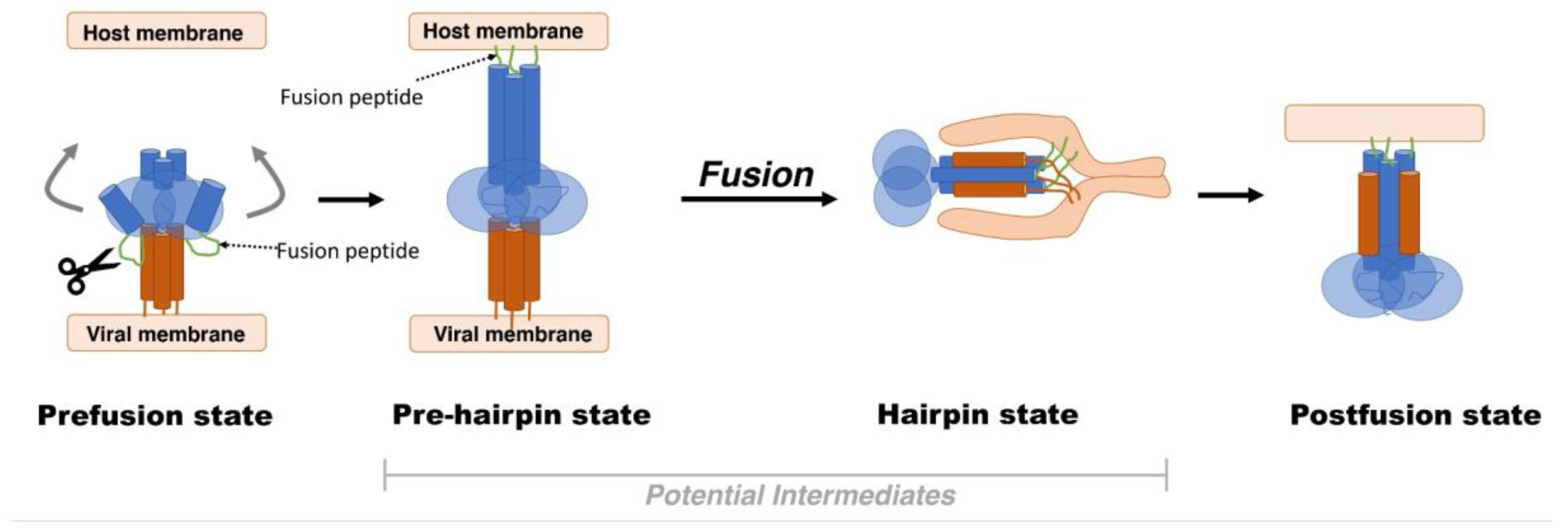
2. Viral Fusion Proteins
3. Antivirals Targeting Viral Surface Proteins
4. Rational Structure-Based Vaccine Design and Development
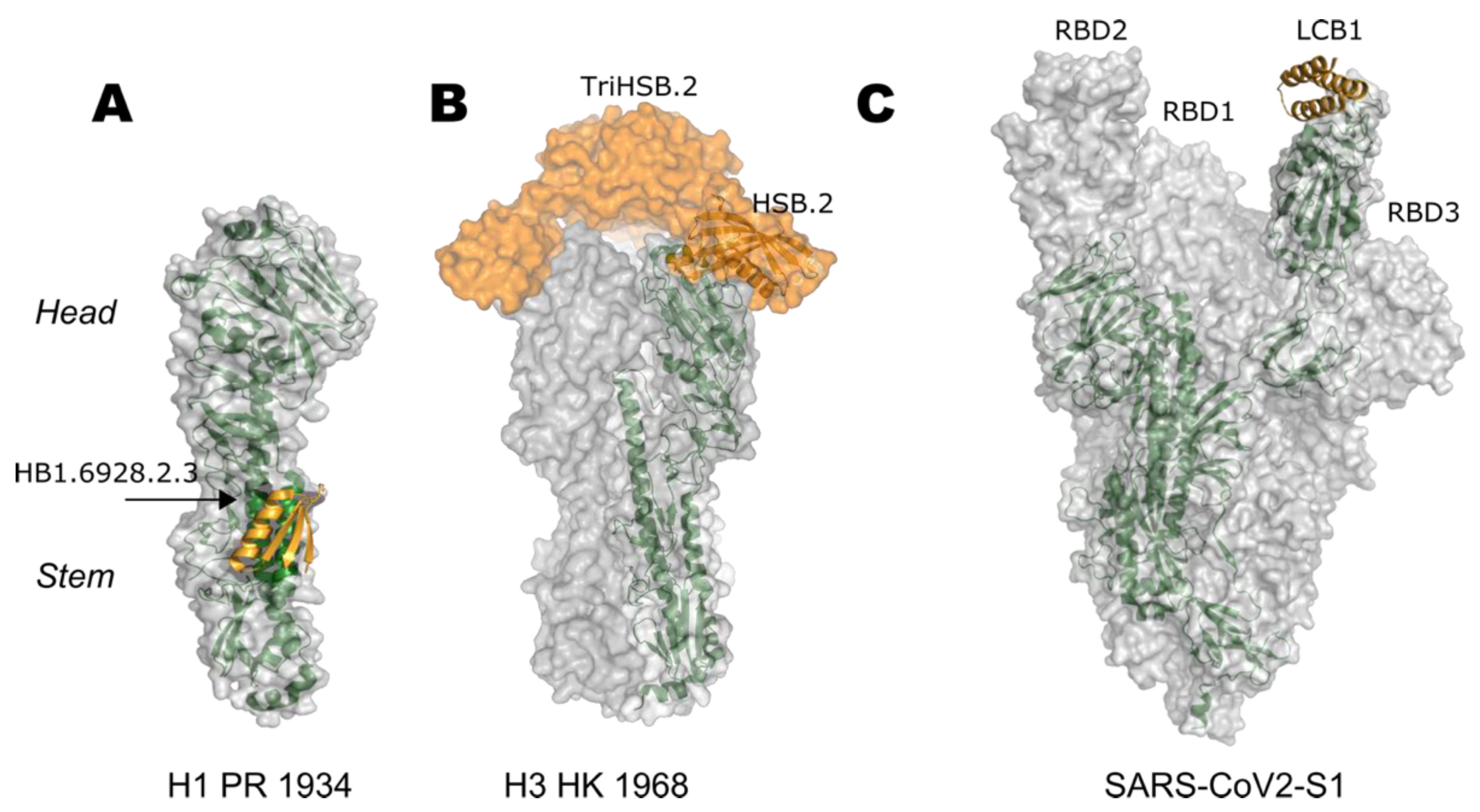
5. Epitope-Focusing through De Novo Scaffolding
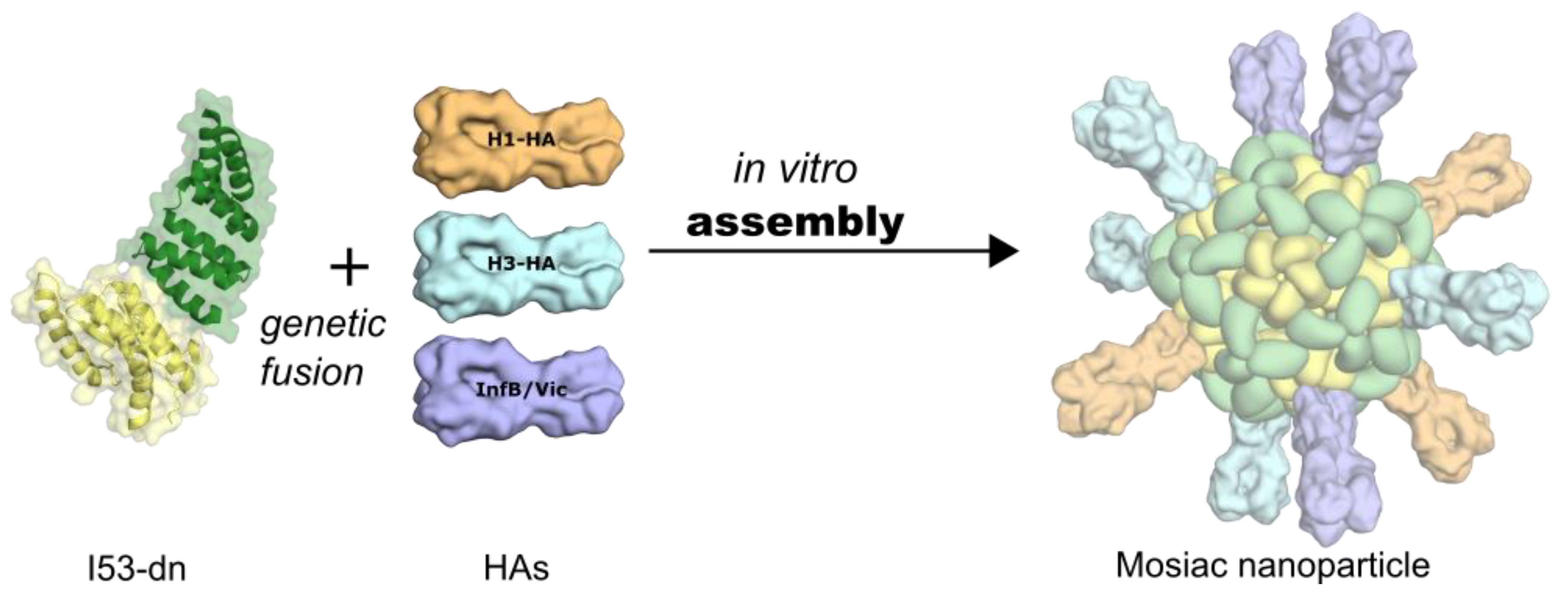
6. Self-Assembling, Designed Nanoparticles for Geometric-Defined Oligomeric Display of Antigens
7. Targeting Sequence Shape Shifters: Influenza
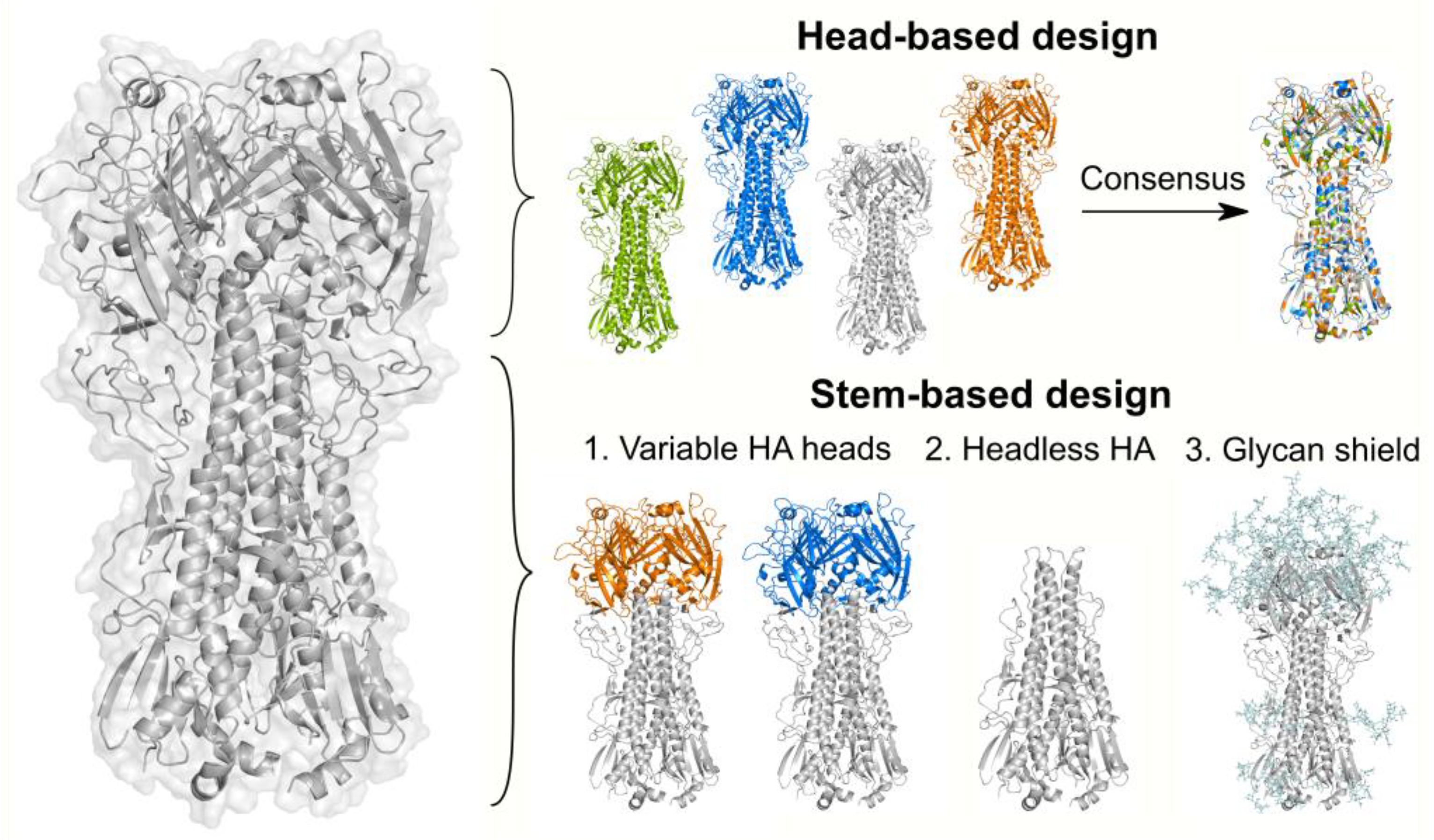
7.1. Head-Based Vaccine Design
7.2. Stem-Based Vaccine Design
7.2.1. Immunization with Antigenically Variable Globular Heads
7.2.2. Removal of the Globular Head
7.2.3. Glycan-Masking of Immunodominant Epitopes
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, A.J.; Armstrong, J.S.; Downie, J.C. From where did the 2009 “swine-origin” influenza A virus (H1N1) emerge? Virol. J. 2009, 6, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Finelli, L.; Shaw, M.W.; Lindstrom, S.; Garten, R.J.; Gubareva, L.V.; Xu, X.; Bridges, C.B.; Uyeki, T.M. Emergence of a Novel Swine-Origin Influenza A (H1N1) Virus in Humans. N. Engl. J. Med. 2009, 360, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Reperant, L.A.; Moesker, F.M.; Osterhaus, A.D.M.E. Influenza: From zoonosis to pandemic. ERJ Open Res. 2016, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lycett, S.J.; Duchatel, F.; Digard, P. A brief history of bird flu. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180257. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, J.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Yu, W.C.; Leung, C.W.; Cheung, C.Y.; Ng, W.F.; Nicholls, J.M.; Ng, T.K.; Chan, K.H.; Lai, S.T.; Lim, W.L.; et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 2004, 363, 617–619. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J. Glob. Health 2019, 9. [Google Scholar] [CrossRef]
- COVID-19 Map—Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 22 March 2021).
- Hu, B.; Zeng, L.-P.; Yang, X.-L.; Ge, X.-Y.; Zhang, W.; Li, B.; Xie, J.-Z.; Shen, X.-R.; Zhang, Y.-Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLOS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.Y.; Jia, N.; Zhang, Y.W.; Shum, M.H.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 972. [Google Scholar] [CrossRef]
- Killerby, M.E.; Biggs, H.M.; Midgley, C.M.; Gerber, S.I.; Watson, J.T. Middle east respiratory syndrome coronavirus transmission. Emerg. Infect. Dis. 2020, 26, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paden, C.R.; Yusof, M.F.B.M.; Al Hammadi, Z.M.; Queen, K.; Tao, Y.; Eltahir, Y.M.; Elsayed, E.A.; Marzoug, B.A.; Bensalah, O.K.A.; Khalafalla, A.I.; et al. Zoonotic origin and transmission of Middle East respiratory syndrome coronavirus in the UAE. Zoonoses Public Health 2018, 65, 322–333. [Google Scholar] [CrossRef]
- Wernery, U.; Lau, S.K.P.; Woo, P.C.Y. Middle East respiratory syndrome (MERS) coronavirus and dromedaries. Vet. J. 2017, 220, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Herfst, S.; Schrauwen, E.J.A.; Linster, M.; Chutinimitkul, S.; De Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012, 336, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Imai, M.; Watanabe, T.; Hatta, M.; Das, S.C.; Ozawa, M.; Shinya, K.; Zhong, G.; Hanson, A.; Katsura, H.; Watanabe, S.; et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 2012, 486, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Bedford, T.; Greninger, A.L.; Roychoudhury, P.; Starita, L.M.; Famulare, M.; Huang, M.L.; Nalla, A.; Pepper, G.; Reinhardt, A.; Xie, H.; et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 2020, 370, 571–575. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef]
- Understanding mRNA COVID-19 Vaccines | CDC. Available online: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines/mrna.html (accessed on 11 May 2021).
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Oliver, S.E.; Gargano, J.W.; Marin, M.; Wallace, M.; Curran, K.G.; Chamberland, M.; McClung, N.; Campos-Outcalt, D.; Morgan, R.L.; Mbaeyi, S.; et al. The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Pfizer-BioNTech COVID-19 Vaccine—United States, December 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 1922–1924. [Google Scholar] [CrossRef]
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.L.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.; Lutrick, K.; et al. Interim Estimates of Vaccine Effectiveness of BNT162b2 and mRNA-1273 COVID-19 Vaccines in Preventing SARS-CoV-2 Infection Among Health Care Personnel, First Responders, and Other Essential and Frontline Workers—Eight U.S. Locations, December 2020–March. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV - A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Siddiqui, P.; Jiang, S. Inactivated SARS-CoV vaccine elicits high titers of spike protein-specific antibodies that block receptor binding and virus entry. Biochem. Biophys. Res. Commun. 2004, 325, 445–452. [Google Scholar] [CrossRef]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e17. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [Green Version]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.E.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-Based Design of a Fusion Glycoprotein Vaccine for Respiratory Syncytial Virus. Science 2013, 342, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widjojoatmodjo, M.N.; Bogaert, L.; Meek, B.; Zahn, R.; Vellinga, J.; Custers, J.; Serroyen, J.; Radošević, K.; Schuitemaker, H. Recombinant low-seroprevalent adenoviral vectors Ad26 and Ad35 expressing the respiratory syncytial virus (RSV) fusion protein induce protective immunity against RSV infection in cotton rats. Vaccine 2015, 33, 5406–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart-Jones, G.B.E.; Chuang, G.Y.; Xu, K.; Zhou, T.; Acharya, P.; Tsybovsky, Y.; Ou, L.; Zhang, B.; Fernandez-Rodriguez, B.; Gilardi, V.; et al. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc. Natl. Acad. Sci. USA 2018, 115, 12265–12270. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Albertini, A.; Bressanelli, S.; Lepault, J.; Gaudin, Y. Structure and Working of Viral Fusion Machinery; Academic Press Inc.: Cambridge, MA, USA, 2011; Volume 68. [Google Scholar]
- Schibli, D.J.; Weissenhorn, W. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 2004, 21, 361–371. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M. Class II virus membrane fusion proteins. Virology 2006, 344, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbilewicz, B. Virus and Cell Fusion Mechanisms. Annu. Rev. Cell Dev. Biol 2014, 30, 111–139. [Google Scholar] [CrossRef] [Green Version]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudin, Y. Reversibility in Fusion Protein Conformational Changes The Intriguing Case of Rhabdovirus-Induced Membrane Fusion. In Fusion of Biological Membranes and Related Problems; Kluwer Academic Publishers: Boston, MA, USA, 2000; pp. 379–408. [Google Scholar]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izaguirre, G. The Proteolytic Regulation of Virus Cell Entry by Furin and Other Proprotein Convertases. Viruses 2019, 11, 837. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef]
- Skehel, J.J.; Bayley, P.M.; Brown, E.B.; Martin, S.R.; Waterfield, M.D.; White, J.M.; Wilson, I.A.; Wiley, D.C. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus mediated membrane fusion. Proc. Natl. Acad. Sci. USA 1982, 79, 968–972. [Google Scholar] [CrossRef] [Green Version]
- Ruigrok, R.W.H.; Martin, S.R.; Wharton, S.A.; Skehel, J.J.; Bayley, P.M.; Wiley, D.C. Conformational changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology 1986, 155, 484–497. [Google Scholar] [CrossRef]
- Chen, J.; Wharton, S.A.; Weissenhorn, W.; Calder, L.J.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA 1995, 92, 12205–12209. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef] [Green Version]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef]
- Muñoz-Barroso, I.; Salzwedel, K.; Hunter, E.; Blumenthal, R. Role of the Membrane-Proximal Domain in the Initial Stages of Human Immunodeficiency Virus Type 1 Envelope Glycoprotein-Mediated Membrane Fusion. J. Virol. 1999, 73, 6089–6092. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.; Greenwell, T.; Matthews, T. A Synthetic Peptide from HIV-1 gp41 is a Potent Inhibitor of Virus-Mediated Cell—Cell Fusion. AIDS Res. Hum. Retrovir. 1993, 9, 1051–1053. [Google Scholar] [CrossRef]
- Leikina, E.; Ramos, C.; Markovic, I.; Zimmerberg, J.; Chernomordik, L.V. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002, 21, 5701–5710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissenhorn, W.; Hinz, A.; Gaudin, Y. Virus membrane fusion. FEBS Lett. 2007, 581, 2150–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O’Neil, R.E.; Faynboym, A.M.; et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 2012, 489, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Yoshida, R.; Ekiert, D.C.; Sakai, N.; Suzuki, Y.; Takada, A.; Wilson, I.A. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. USA 2012, 109, 17040–17045. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.S.; Ohshima, N.; Stanfield, R.L.; Yu, W.; Iba, Y.; Okuno, Y.; Kurosawa, Y.; Wilson, I.A. Receptor mimicry by antibody F045-092 facilitates universal binding to the H3 subtype of influenza virus. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, L.M.; Patel, A. Timely antiviral administration during an influenza pandemic: Key components. Am. J. Public Health 2018, 108, S215–S220. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Mitoma, F.; Sibbald, R.G.; Shafran, S.D.; Boon, R.; Saltzman, R.L. Oral famciclovir for the suppression of recurrent genital herpes: A randomized controlled trial. J. Am. Med. Assoc. 1998, 280, 887–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, A.S.F.; McMahon, B.J. Chronic hepatitis B: Update of recommendations. Hepatology 2004, 39, 857–861. [Google Scholar] [CrossRef]
- Olchanski, N.; Hansen, R.N.; Pope, E.; D’Cruz, B.; Fergie, J.; Goldstein, M.; Krilov, L.R.; McLaurin, K.K.; Nabrit-Stephens, B.; Oster, G.; et al. Palivizumab prophylaxis for respiratory syncytial virus: Examining the evidence around value. Open Forum Infect. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Su, S.; Borné, Y. A meta-analysis of the efficacy of HAART on HIV transmission and its impact on sexual risk behaviours among men who have sex with men. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Collier, A.C.; Coombs, R.W.; Schoenfeld, D.A.; Bassett, R.L.; Timpone, J.; Baruch, A.; Jones, M.; Facey, K.; Whitacre, C.; McAuliffe, V.J.; et al. Treatment of Human Immunodeficiency Virus Infection with Saquinavir, Zidovudine, and Zalcitabine. N. Engl. J. Med. 1996, 334, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Grobler, J.A.; Anderson, A.S.; Fernandes, P.; Diamond, M.S.; Colvis, C.M.; Menetski, J.P.; Alvarez, R.M.; Young, J.A.T.; Carter, K.L. Accelerated Preclinical Paths to Support Rapid Development of COVID-19 Therapeutics. Cell Host Microbe 2020, 28, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.; Chan-Tack, K.; Farley, J.; Sherwat, A. FDA Approval of Remdesivir—A Step in the Right Direction. N. Engl. J. Med. 2020, 383, 2598–2600. [Google Scholar] [CrossRef]
- FDA Approves First Treatment for COVID-19 | FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 29 March 2021).
- Zaraket, H.; Saito, R. Japanese Surveillance Systems and Treatment for Influenza. Curr. Treat. Options Infect. Dis. 2016, 8, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Selisko, B.; Le, N.T.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Nirula, A.; Chen, P.; Boscia, J.; Heller, B.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19. JAMA 2021, 325, 632. [Google Scholar] [CrossRef]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 229–237. [Google Scholar] [CrossRef]
- New Data Show Treatment with Lilly’s Neutralizing Antibodies Bamlanivimab (LY-CoV555) and Etesevimab (LY-CoV016) Together Reduced risk of COVID-19 Hospitalizations and Death by 70 Percent | Eli Lilly and Company. Available online: https://investor.lilly.com/news-releases/news-release-details/new-data-show-treatment-lillys-neutralizing-antibodies (accessed on 29 March 2021).
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Fleishman, S.J.; Whitehead, T.A.; Ekiert, D.C.; Dreyfus, C.; Corn, J.E.; Strauch, E.M.; Wilson, I.A.; Baker, D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 2011, 332, 816–821. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, A.; Silva, D.A.; Rocklin, G.J.; Hicks, D.R.; Vergara, R.; Murapa, P.; Bernard, S.M.; Zhang, L.; Lam, K.H.; Yao, G.; et al. Massively parallel de novo protein design for targeted therapeutics. Nature 2017, 550, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Friesen, R.H.E.; Bhabha, G.; Kwaks, T.; Jongeneelen, M.; Yu, W.; Ophorst, C.; Cox, F.; Korse, H.J.W.M.; Brandenburg, B.; et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 2011, 333, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauch, E.M.; Bernard, S.M.; La, D.; Bohn, A.J.; Lee, P.S.; Anderson, C.E.; Nieusma, T.; Holstein, C.A.; Garcia, N.K.; Hooper, K.A.; et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 2017, 35, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Bricker, T.L.; Darling, T.L.; Hassan, A.O.; Harastani, H.H.; Soung, A.; Jiang, X.; Dai, Y.N.; Zhao, H.; Adams, L.J.; Holtzman, M.J.; et al. A single intranasal or intramuscular immunization with chimpanzee adenovirus vectored SARS-CoV-2 vaccine protects against pneumonia in hamsters. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liang, B.; Surman, S.; Amaro-Carambot, E.; Kabatova, B.; Mackow, N.; Lingemann, M.; Yang, L.; McLellan, J.S.; Graham, B.S.; Kwong, P.D.; et al. Enhanced Neutralizing Antibody Response Induced by Respiratory Syncytial Virus Prefusion F Protein Expressed by a Vaccine Candidate. J. Virol. 2015, 89, 9499–9510. [Google Scholar] [CrossRef] [Green Version]
- Rossey, I.; McLellan, J.S.; Saelens, X.; Schepens, B. Clinical Potential of Prefusion RSV F-specific Antibodies. Trends Microbiol. 2018, 26, 209–219. [Google Scholar] [CrossRef]
- Falloon, J.; Yu, J.; Esser, M.T.; Villafana, T.; Yu, L.; Dubovsky, F.; Takas, T.; Levin, M.J.; Falsey, A.R. An Adjuvanted, Postfusion F Protein–Based Vaccine Did Not Prevent Respiratory Syncytial Virus Illness in Older Adults. J. Infect. Dis. 2017, 216, 1362–1370. [Google Scholar] [CrossRef]
- Graham, B.S. Vaccine development for respiratory syncytial virus. Curr. Opin. Virol. 2017, 23, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.Y.; et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef] [Green Version]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Cullen, L.M.G.; Blanco, J.C.G.; Morrison, T.G. Cotton rat immune responses to virus-like particles containing the pre-fusion form of respiratory syncytial virus fusion protein. J. Transl. Med. 2015, 13, 350. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.P.; Khan, A.A.; Esser, M.T.; Jensen, K.; Takas, T.; Kankam, M.K.; Villafana, T.; Dubovsky, F. Safety, tolerability, and pharmacokinetics of MEDI8897, the respiratory syncytial virus prefusion F-targeting monoclonal antibody with an extended half-life, in healthy adults. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krarup, A.; Truan, D.; Furmanova-Hollenstein, P.; Bogaert, L.; Bouchier, P.; Bisschop, I.J.M.; Widjojoatmodjo, M.N.; Zahn, R.; Schuitemaker, H.; McLellan, J.S.; et al. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat. Commun. 2015, 6, 8143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battles, M.B.; Más, V.; Olmedillas, E.; Cano, O.; Vázquez, M.; Rodríguez, L.; Melero, J.A.; McLellan, J.S. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Juraszek, J.; Rutten, L.; Blokland, S.; Bouchier, P.; Voorzaat, R.; Ritschel, T.; Bakkers, M.J.G.; Renault, L.L.R.; Langedijk, J.P.M. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.W.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 Spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Zhang, L.; Durr, E.; Galli, J.D.; Cosmi, S.; Cejas, P.J.; Luo, B.; Touch, S.; Parmet, P.; Fridman, A.; Espeseth, A.S.; et al. Design and characterization of a fusion glycoprotein vaccine for Respiratory Syncytial Virus with improved stability. Vaccine 2018, 36, 8119–8130. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Joyce, M.G.; Zhang, B.; Ou, L.; Chen, M.; Chuang, G.Y.; Druz, A.; Kong, W.P.; Lai, Y.T.; Rundlet, E.J.; Tsybovsky, Y.; et al. Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat. Struct. Mol. Biol. 2016, 23, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Stewart-Jones, G.B.E.; Thomas, P.V.; Chen, M.; Druz, A.; Joyce, M.G.; Kong, W.P.; Sastry, M.; Soto, C.; Yang, Y.; Zhang, B.; et al. A cysteine zipper stabilizes a pre-fusion F glycoprotein vaccine for respiratory syncytial virus. PLoS ONE 2015, 10, e0128779. [Google Scholar] [CrossRef]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef]
- Correia, B.E.; Ban, Y.E.A.; Holmes, M.A.; Xu, H.; Ellingson, K.; Kraft, Z.; Carrico, C.; Boni, E.; Sather, D.N.; Zenobia, C.; et al. Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 2010, 18, 1116–1126. [Google Scholar] [CrossRef] [Green Version]
- Ofek, G.; Guenaga, F.J.; Schief, W.R.; Skinner, J.; Baker, D.; Wyatt, R.; Kwong, P.D. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 2010, 107, 17880–17887. [Google Scholar] [CrossRef] [Green Version]
- McLellan, J.S.; Correia, B.E.; Chen, M.; Yang, Y.; Graham, B.S.; Schief, W.R.; Kwong, P.D. Design and characterization of epitope-scaffold immunogens that present the motavizumab epitope from respiratory syncytial virus. J. Mol. Biol. 2011, 409, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, M.L.; Correia, B.E.; Ban, Y.E.A.; Carrico, C.; Kalyuzhniy, O.; Chen, L.; Schroeter, A.; Huang, P.S.; McLellan, J.S.; Kwong, P.D.; et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 2011, 334, 373–376. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, M.L.; Ban, Y.E.A.; Julien, J.P.; Bryson, S.; Schroeter, A.; Kalyuzhniy, O.; Porter, J.R.; Adachi, Y.; Baker, D.; Pai, E.F.; et al. Computational design of high-affinity epitope scaffolds by backbone grafting of a linear epitope. J. Mol. Biol. 2012, 415, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pfarr, D.S.; Johnson, S.; Brewah, Y.A.; Woods, R.M.; Patel, N.K.; White, W.I.; Young, J.F.; Kiener, P.A. Development of Motavizumab, an Ultra-potent Antibody for the Prevention of Respiratory Syncytial Virus Infection in the Upper and Lower Respiratory Tract. J. Mol. Biol. 2007, 368, 652–665. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Kim, A.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structural basis of respiratory syncytial virus neutralization by motavizumab. Nat. Struct. Mol. Biol. 2010, 17, 248–250. [Google Scholar] [CrossRef]
- Sesterhenn, F.; Yang, C.; Bonet, J.; Cramer, J.T.; Wen, X.; Wang, Y.; Chiang, C.I.; Abriata, L.A.; Kucharska, I.; Castoro, G.; et al. De novo protein design enables the precise induction of RSV-neutralizing antibodies. Science 2020, 368. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Chang, J.-S.; Yang, Y.; Kim, A.; Graham, B.S.; Kwong, P.D. Structure of a Major Antigenic Site on the Respiratory Syncytial Virus Fusion Glycoprotein in Complex with Neutralizing Antibody 101F. J. Virol. 2010, 84, 12236–12244. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Liu, J.; Bess, J.; Chertova, E.; Lifson, J.D.; Grisé, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Vahey, M.D.; Fletcher, D.A. Influenza A virus surface proteins are organized to help penetrate host mucus. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Wei, C.J.; Yassine, H.M.; McTamney, P.M.; Boyington, J.C.; Whittle, J.R.R.; Rao, S.S.; Kong, W.P.; Wang, L.; Nabel, G.J. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 2013, 499, 102–106. [Google Scholar] [CrossRef]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [CrossRef] [PubMed]
- King, N.P.; Bale, J.B.; Sheffler, W.; McNamara, D.E.; Gonen, S.; Gonen, T.; Yeates, T.O.; Baker, D. Accurate design of co-assembling multi-component protein nanomaterials. Nature 2014, 510, 103–108. [Google Scholar] [CrossRef]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Ueda, G.; Antanasijevic, A.; Fallas, J.A.; Sheffler, W.; Copps, J.; Ellis, D.; Hutchinson, G.B.; Moyer, A.; Yasmeen, A.; Tsybovsky, Y.; et al. Tailored design of protein nanoparticle scaffolds for multivalent presentation of viral glycoprotein antigens. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Fiala, B.; Schäfer, A.; Wrenn, S.; Pham, M.N.; Murphy, M.; Tse, L.V.; Shehata, L.; O’Connor, M.A.; Chen, C.; et al. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 2020, 183, 1367–1382.e17. [Google Scholar] [CrossRef]
- Boyoglu-Barnum, S.; Ellis, D.; Gillespie, R.A.; Hutchinson, G.B.; Park, Y.-J.; Moin, S.M.; Acton, O.J.; Ravichandran, R.; Murphy, M.; Pettie, D.; et al. Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature 2021, 1–6. [Google Scholar] [CrossRef]
- Schweiger, B.; Zadow, I.; Heckler, R. Antigenic drift and variability of influenza viruses. Med. Microbiol. Immunol. 2002, 191, 133–138. [Google Scholar] [CrossRef]
- Wei, C.J.; Crank, M.C.; Shiver, J.; Graham, B.S.; Mascola, J.R.; Nabel, G.J. Next-generation influenza vaccines: Opportunities and challenges. Nat. Rev. Drug Discov. 2020, 19, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Fiore, A.E.; Bridges, C.B.; Cox, N.J. Seasonal influenza vaccines. Curr. Top. Microbiol. Immunol. 2009, 333, 43–82. [Google Scholar] [CrossRef]
- McMillan, C.L.D.; Young, P.R.; Watterson, D.; Chappell, K.J. The next generation of influenza vaccines: Towards a universal solution. Vaccines 2021, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: Different approaches for one goal. Virol. J. 2018, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hiroi, S.; Kuhara, M.; Kishi, Y.; Ono, K.i.; Matsuzawa, S.; Yamamoto, N.; Komano, J. Human monoclonal antibodies neutralizing influenza virus A/H1N1pdm09 and seasonal A/H1N1 strains—Distinct Ig gene repertoires with a similar action mechanism. Immunobiology 2018, 223, 319–326. [Google Scholar] [CrossRef]
- Giles, B.M.; Ross, T.M. A computationally optimized broadly reactive antigen (COBRA) based H5N1 VLP vaccine elicits broadly reactive antibodies in mice and ferrets. Vaccine 2011, 29, 3043–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, D.M.; Darby, C.A.; Lefoley, B.C.; Crevar, C.J.; Alefantis, T.; Oomen, R.; Anderson, S.F.; Strugnell, T.; Cortés-Garcia, G.; Vogel, T.U.; et al. Design and Characterization of a Computationally Optimized Broadly Reactive Hemagglutinin Vaccine for H1N1 Influenza Viruses. J. Virol. 2016, 90, 4720–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.M.; Allen, J.D.; Bebin-Blackwell, A.-G.; Carter, D.M.; Alefantis, T.; DiNapoli, J.; Kleanthous, H.; Ross, T.M. Computationally Optimized Broadly Reactive Hemagglutinin Elicits Hemagglutination Inhibition Antibodies against a Panel of H3N2 Influenza Virus Cocirculating Variants. J. Virol. 2017, 91, 1581–1598. [Google Scholar] [CrossRef] [Green Version]
- Bohne-Lang, A.; Von der Lieth, C.W. GlyProt: In silico glycosylation of proteins. Nucleic Acids Res. 2005, 33, W214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System; Version 1.8; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Hong, M.; Lee, P.S.; Hoffman, R.M.B.; Zhu, X.; Krause, J.C.; Laursen, N.S.; Yoon, S.-i.; Song, L.; Tussey, L.; Crowe, J.E.; et al. Antibody Recognition of the Pandemic H1N1 Influenza Virus Hemagglutinin Receptor Binding Site. J. Virol. 2013, 87, 12471–12480. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.B.; Van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L.L.M.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al. Heterosubtypic Neutralizing Monoclonal Antibodies Cross-Protective against H5N1 and H1N1 Recovered from Human IgM+ Memory B Cells. PLoS ONE 2008, 3, e3942. [Google Scholar] [CrossRef] [Green Version]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.A.; Friesen, R.H.E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ellebedy, A.H.; Krammer, F.; Li, G.M.; Miller, M.S.; Chiu, C.; Wrammert, J.; Chang, C.Y.; Davis, C.W.; McCausland, M.; Elbein, R.; et al. Induction of broadly cross-reactive antibody responses to the influenza HA stem region following H5N1 vaccination in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 13133–13138. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Pica, N.; Hai, R.; Tan, G.S.; Palese, P. Hemagglutinin Stalk-Reactive Antibodies Are Boosted following Sequential Infection with Seasonal and Pandemic H1N1 Influenza Virus in Mice. J. Virol. 2012, 86, 10302–10307. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.S.; Tsibane, T.; Krammer, F.; Hai, R.; Rahmat, S.; Basler, C.F.; Palese, P. 1976 and 2009 H1N1 Influenza Virus Vaccines Boost Anti-Hemagglutinin Stalk Antibodies in Humans. J. Infect. Dis. 2013, 207, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Sui, J.; Sheehan, J.; Hwang, W.C.; Bankston, L.A.; Burchett, S.K.; Huang, C.Y.; Liddington, R.C.; Beigel, J.H.; Marasco, W.A. Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza a antibodies. Clin. Infect. Dis. 2011, 52, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, K.E.; Henry Dunand, C.J.; Wilson, P.C. Heads, stalks and everything else: How can antibodies eradicate influenza as a human disease? Curr. Opin. Immunol. 2016, 42, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Nachbagauer, R.; Kinzler, D.; Choi, A.; Hirsh, A.; Beaulieu, E.; Lecrenier, N.; Innis, B.L.; Palese, P.; Mallett, C.P.; Krammer, F. A chimeric haemagglutinin-based influenza split virion vaccine adjuvanted with AS03 induces protective stalk-reactive antibodies in mice. Jpn. Vaccines 2016, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Ermler, M.E.; Kirkpatrick, E.; Sun, W.; Hai, R.; Amanat, F.; Chromikova, V.; Palese, P.; Krammer, F. Chimeric Hemagglutinin Constructs Induce Broad Protection against Influenza B Virus Challenge in the Mouse Model. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Pica, N.; Hai, R.; Margine, I.; Palese, P. Chimeric Hemagglutinin Influenza Virus Vaccine Constructs Elicit Broadly Protective Stalk-Specific Antibodies. J. Virol. 2013, 87, 6542–6550. [Google Scholar] [CrossRef] [Green Version]
- Hai, R.; Krammer, F.; Tan, G.S.; Pica, N.; Eggink, D.; Maamary, J.; Margine, I.; Albrecht, R.A.; Palese, P. Influenza Viruses Expressing Chimeric Hemagglutinins: Globular Head and Stalk Domains Derived from Different Subtypes. J. Virol. 2012, 86, 5774–5781. [Google Scholar] [CrossRef] [Green Version]
- Nachbagauer, R.; Feser, J.; Naficy, A.; Bernstein, D.I.; Guptill, J.; Walter, E.B.; Berlanda-Scorza, F.; Stadlbauer, D.; Wilson, P.C.; Aydillo, T.; et al. A chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase I trial. Nat. Med. 2021, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Liu, S.T.H.; Suntronwong, N.; Sun, W.; Bailey, M.J.; Nachbagauer, R.; Krammer, F.; Palese, P. A mosaic hemagglutinin-based influenza virus vaccine candidate protects mice from challenge with divergent H3N2 strains. Jpn. Vaccines 2019, 4, 1–9. [Google Scholar] [CrossRef]
- Sun, W.; Kirkpatrick, E.; Ermler, M.; Nachbagauer, R.; Broecker, F.; Krammer, F.; Palese, P. Development of Influenza B Universal Vaccine Candidates Using the “Mosaic” Hemagglutinin Approach. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, F.; Palese, P. Universal Influenza Virus Vaccines That Target the Conserved Hemagglutinin Stalk and Conserved Sites in the Head Domain. J. Infect. Dis. 2019, 219, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, S.A.; Mallajosyula, V.V.A.; Li, O.T.W.; Chin, A.W.H.; Carnell, G.; Temperton, N.; Varadarajan, R.; Poon, L.L.M. Stalking influenza by vaccination with pre-fusion headless HA mini-stem. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Welsh, J.P.; Swartz, J.R. Production and stabilization of the trimeric influenza hemagglutinin stem domain for potentially broadly protective influenza vaccines. Proc. Natl. Acad. Sci. USA 2014, 111, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, K.S.; Moin, S.M.; Yassine, H.M.; Cagigi, A.; Kanekiyo, M.; Boyoglu-Barnum, S.; Myers, S.I.; Tsybovsky, Y.; Wheatley, A.K.; Schramm, C.A.; et al. Design of nanoparticulate group 2 influenza virus hemagglutinin stem antigens that activate unmutated ancestor B cell receptors of broadly neutralizing antibody lineages. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steel, J.; Lowen, A.C.; Wang, T.T.; Yondola, M.; Gao, Q.; Haye, K.; García-Sastre, A.; Palesea, P. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. MBio 2010, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommakanti, G.; Lu, X.; Citron, M.P.; Najar, T.A.; Heidecker, G.J.; ter Meulen, J.; Varadarajan, R.; Liang, X. Design of Escherichia coli-Expressed Stalk Domain Immunogens of H1N1 Hemagglutinin That Protect Mice from Lethal Challenge. J. Virol. 2012, 86, 13434–13444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallajosyula, V.V.A.; Citron, M.; Ferrara, F.; Lu, X.; Callahan, C.; Heidecker, G.J.; Sarma, S.P.; Flynn, J.A.; Temperton, N.J.; Liang, X.; et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc. Natl. Acad. Sci. USA 2014, 111, E2514–E2523. [Google Scholar] [CrossRef] [Green Version]
- Mallajosyula, V.V.A.; Citron, M.; Ferrara, F.; Temperton, N.J.; Liang, X.; Flynn, J.A.; Varadarajan, R. Hemagglutinin sequence conservation guided stem immunogen design from influenza A H3 subtype. Front. Immunol. 2015, 6, 329. [Google Scholar] [CrossRef] [Green Version]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.J.; Kanekiyo, M.; Kong, W.P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Wohlbold, T.J.; Nachbagauer, R.; Margine, I.; Tan, G.S.; Hirsh, A.; Krammer, F. Vaccination with soluble headless hemagglutinin protects mice from challenge with divergent influenza viruses. Vaccine 2015, 33, 3314–3321. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C.; Liao, H.Y.; Zhang, J.Y.; Cheng, T.J.R.; Wong, C.H. Development of a universal influenza vaccine using hemagglutinin stem protein produced from Pichia pastoris. Virology 2019, 526, 125–137. [Google Scholar] [CrossRef]
- National Institute of Allergy and Infectious Diseases (NIAID) Influenza HA Ferritin Vaccine, Alone or in Prime-Boost Regimens With an Influenza DNA Vaccine in Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT03186781 (accessed on 6 June 2021).
- Lin, S.-C.; Lin, Y.-F.; Chong, P.; Wu, S.-C. Broader Neutralizing Antibodies against H5N1 Viruses Using Prime-Boost Immunization of Hyperglycosylated Hemagglutinin DNA and Virus-Like Particles. PLoS ONE 2012, 7, e39075. [Google Scholar] [CrossRef]
- Lin, S.-C.; Liu, W.-C.; Jan, J.-T.; Wu, S.-C. Glycan Masking of Hemagglutinin for Adenovirus Vector and Recombinant Protein Immunizations Elicits Broadly Neutralizing Antibodies against H5N1 Avian Influenza Viruses. PLoS ONE 2014, 9, e92822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajic, G.; Maron, M.J.; Adachi, Y.; Onodera, T.; McCarthy, K.R.; McGee, C.E.; Sempowski, G.D.; Takahashi, Y.; Kelsoe, G.; Kuraoka, M.; et al. Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe 2019, 25, 827–835.e6. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the Immune Response against Influenza Virus Hemagglutinin toward the Conserved Stalk Domain by Hyperglycosylation of the Globular Head Domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.-C.; Jan, J.-T.; Huang, Y.-J.; Chen, T.-H.; Wu, S.-C. Unmasking Stem-Specific Neutralizing Epitopes by Abolishing N-Linked Glycosylation Sites of Influenza Virus Hemagglutinin Proteins for Vaccine Design. J. Virol. 2016, 90, 8496–8508. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narkhede, Y.B.; Gonzalez, K.J.; Strauch, E.-M. Targeting Viral Surface Proteins through Structure-Based Design. Viruses 2021, 13, 1320. https://doi.org/10.3390/v13071320
Narkhede YB, Gonzalez KJ, Strauch E-M. Targeting Viral Surface Proteins through Structure-Based Design. Viruses. 2021; 13(7):1320. https://doi.org/10.3390/v13071320
Chicago/Turabian StyleNarkhede, Yogesh B, Karen J Gonzalez, and Eva-Maria Strauch. 2021. "Targeting Viral Surface Proteins through Structure-Based Design" Viruses 13, no. 7: 1320. https://doi.org/10.3390/v13071320
APA StyleNarkhede, Y. B., Gonzalez, K. J., & Strauch, E.-M. (2021). Targeting Viral Surface Proteins through Structure-Based Design. Viruses, 13(7), 1320. https://doi.org/10.3390/v13071320