
Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Phylogeny Reconstruction and Lineage/Sublineage Classification
2.3. Mutation Detection of ORFs
2.4. Identification of Potential Glycosylation Sites in L1 and L2 Proteins
2.5. Nucleotide Composition Analysis
2.6. Analysis of Effective Number of Codons
2.7. Neutrality Plot Analysis
2.8. Codon Usage Frequency Analysis
3. Results
3.1. Classification of HPV16 Lineages and Sublineages
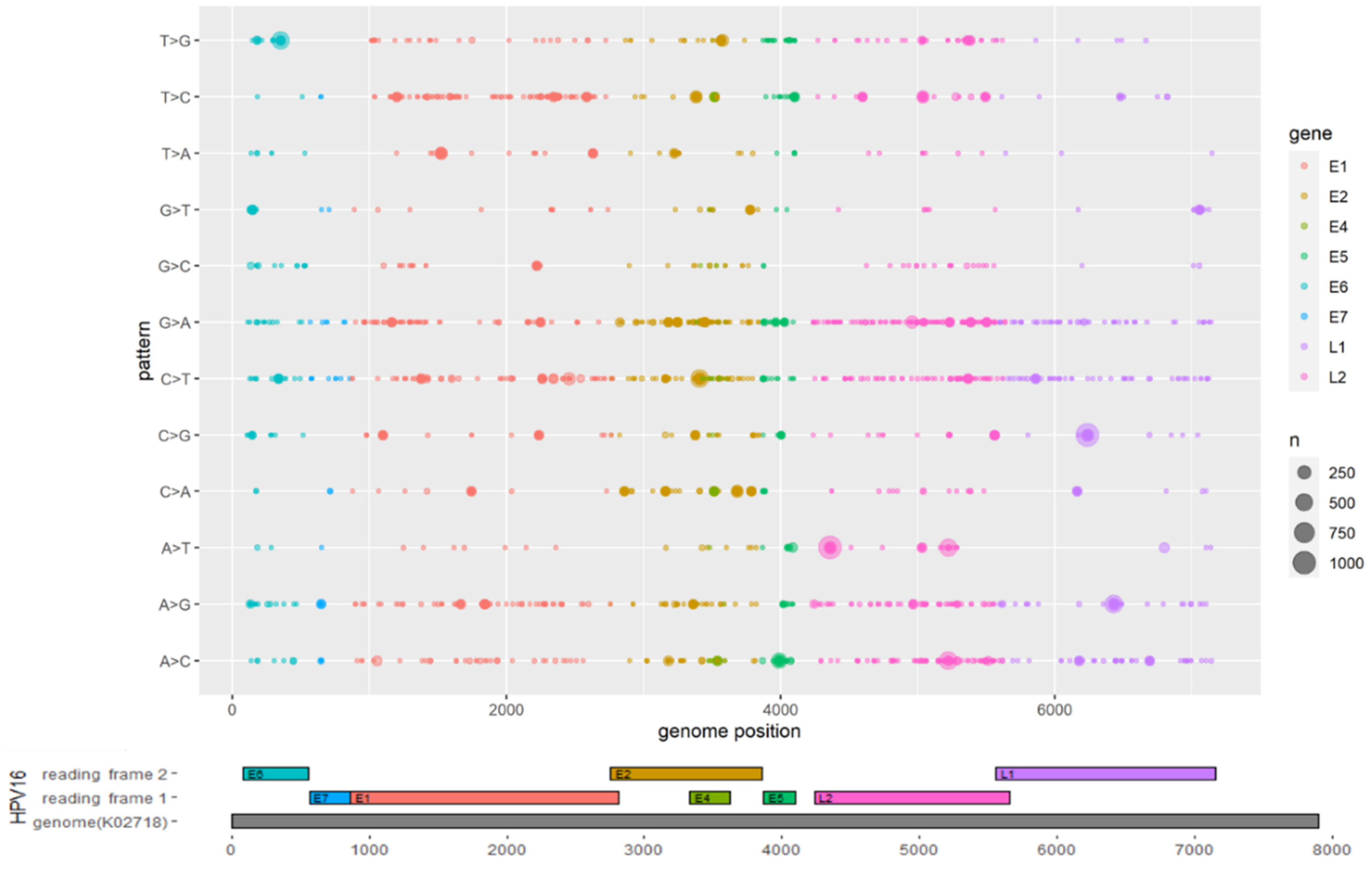
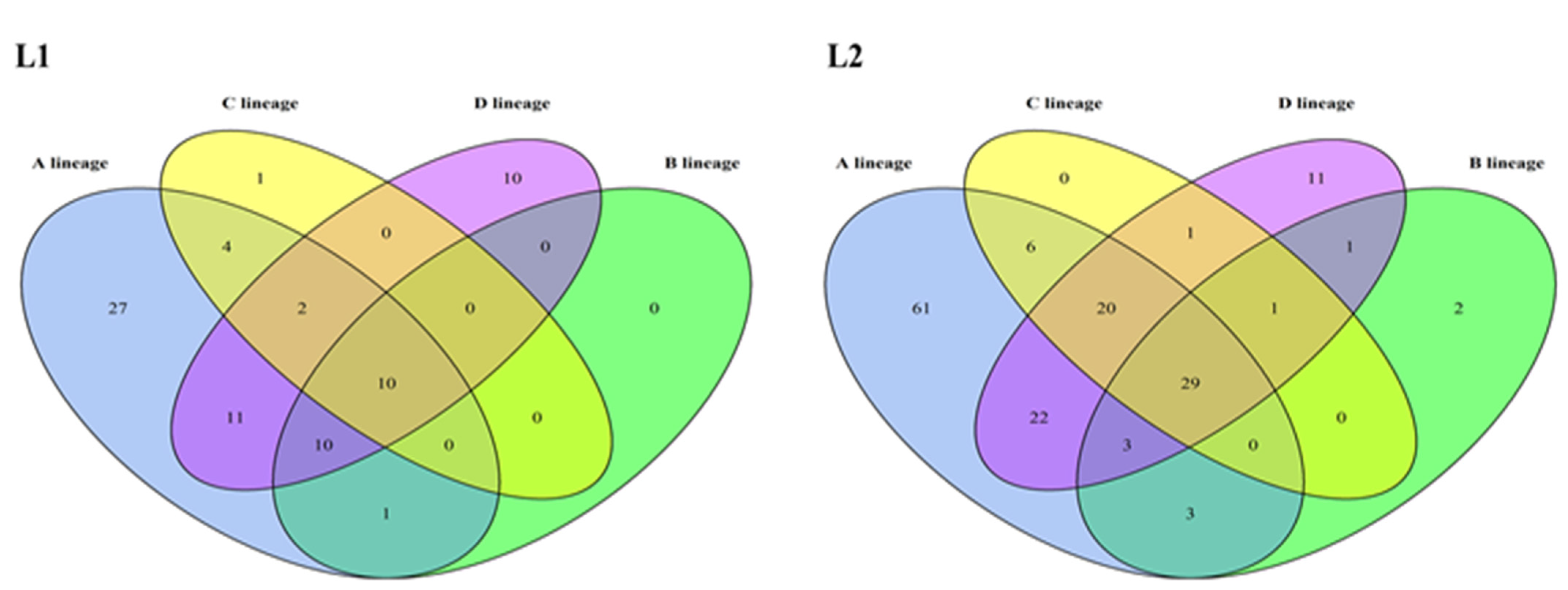
3.2. Mutations Identified across the HPV16 Genome
3.3. Glycosylation Analysis of HPV16 L1 and L2 Proteins
3.4. Nucleotide Composition of HPV16 Genomes
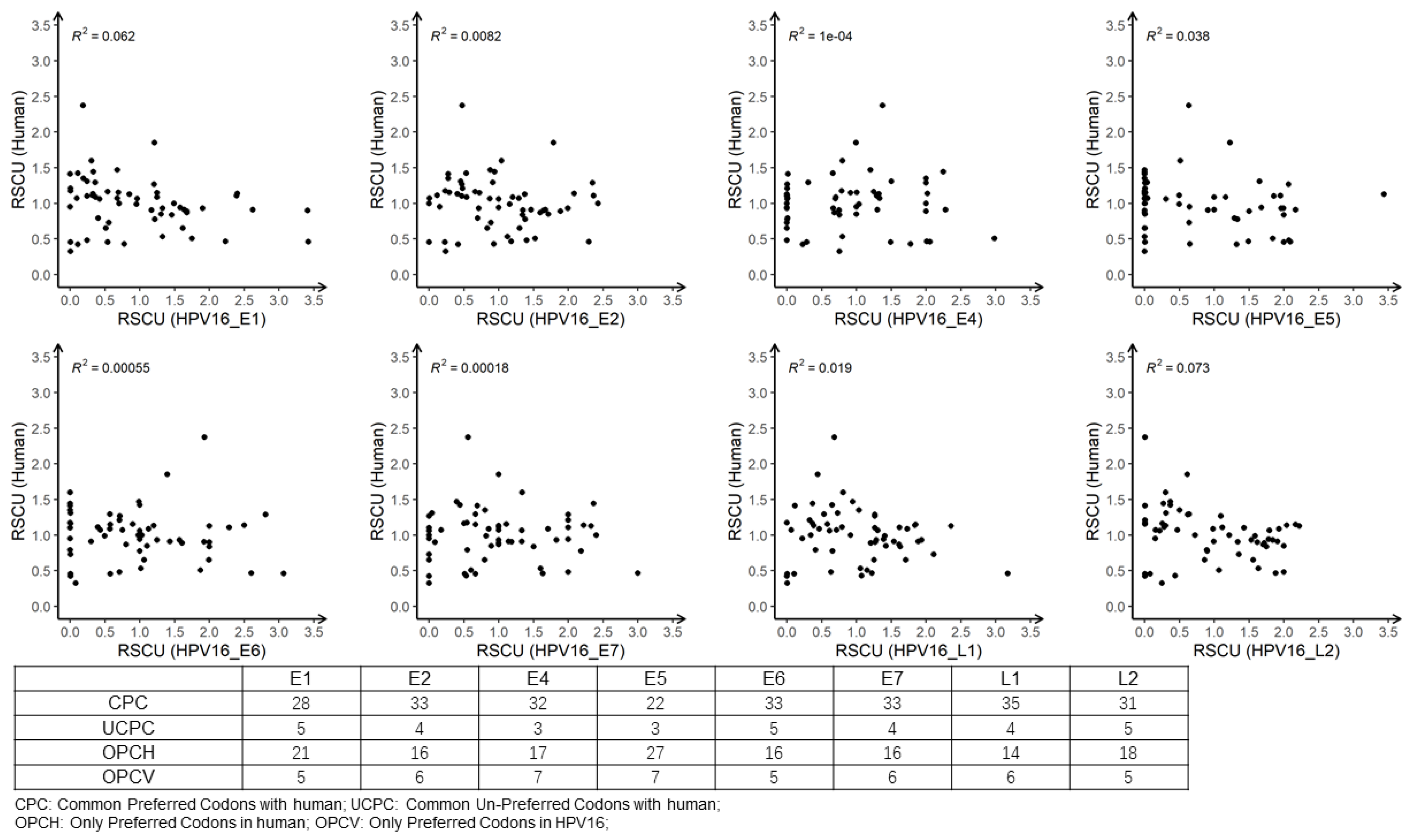
3.5. The Effect of Mutational and Natural Selection Pressure on CUB of HPV16
3.6. Analysis of RSCU
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burd, E.M. Human Papillomavirus and Cervical Cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef]
- Crow, J.M. HPV: The global burden. Nat. Cell Biol. 2012, 488, S2–S3. [Google Scholar] [CrossRef]
- Woodman, C.B.J.; Collins, S.I.; Young, L. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef]
- Fehrmann, F.; Laimins, L.A. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 2003, 22, 5201–5207. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2014, 445, 57–79. [Google Scholar] [CrossRef]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef]
- Pol, S.B.V.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef]
- Yadav, R.; Zhai, L.; Tumban, E. Virus-like Particle-Based L2 Vaccines against HPVs: Where Are We Today? Viruses 2019, 12, 18. [Google Scholar] [CrossRef]
- Farmer, E.; Cheng, M.A.; Hung, C.-F.; Wu, T.-C. Vaccination Strategies for the Control and Treatment of HPV Infection and HPV-Associated Cancer. Methods Mol. Biol. 2021, 217, 157–195. [Google Scholar] [CrossRef]
- de Villiers, E.-M.; Fauquet, C.; Broker, T.R.; Bernard, H.-U.; Hausen, H.Z. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Bzhalava, D.; Eklund, C.; Dillner, J. International standardization and classification of human papillomavirus types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schiffman, M.; Herrero, R.; DeSalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and Taxonomic Classification of Human Papillomavirus 16 (HPV16)-Related Variant Genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef]
- Chen, Z.; De Freitas, L.B.; Burk, R.D. Evolution and Classification of Oncogenic Human Papillomavirus Types and Variants Associated with Cervical Cancer. Methods Mol. Biol. 2015, 1249, 3–26. [Google Scholar] [CrossRef]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef]
- Cornet, I.; Gheit, T.; Franceschi, S.; Vignat, J.; Burk, R.D.; Sylla, B.S.; Tommasino, M.; Clifford, G.M.; the IARC HPV Variant Study Group. Human Papillomavirus Type 16 Genetic Variants: Phylogeny and Classification Based on E6 and LCR. J. Virol. 2012, 86, 6855–6861. [Google Scholar] [CrossRef]
- Park, J.S.; Shin, S.; Kim, E.; Kim, J.E.; Kim, Y.B.; Oh, S.; Roh, E.Y.; Yoon, J.H. Association of human papillomavirus type 16 and its genetic variants with cervical lesion in Korea. APMIS 2016, 124, 950–957. [Google Scholar] [CrossRef]
- Hildesheim, A.; Schiffman, M.; Bromley, C.; Wacholder, S.; Herrero, R.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Scarpidis, U.; Lin, Q.-Q.; et al. Human Papillomavirus Type 16 Variants and Risk of Cervical Cancer. J. Natl. Cancer Inst. 2001, 93, 315–318. [Google Scholar] [CrossRef]
- Rader, J.S.; Tsaih, S.-W.; Fullin, D.; Murray, M.W.; Iden, M.; Zimmermann, M.T.; Flister, M.J. Genetic variations in human papillomavirus and cervical cancer outcomes. Int. J. Cancer 2019, 144, 2206–2214. [Google Scholar] [CrossRef]
- Clifford, G.M.; Tenet, V.; Georges, D.; Alemany, L.; Pavón, M.A.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Bass, S.; et al. Human papillomavirus 16 sub-lineage dispersal and cervical cancer risk worldwide: Whole viral genome sequences from 7116 HPV16-positive women. Papillomavirus Res. 2019, 7, 67–74. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108, 100. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, R.A.; Saunders, C.; Hensel, M.; Stamatatos, L. N-Linked Glycosylation of the V3 Loop and the Immunologically Silent Face of gp120 Protects Human Immunodeficiency Virus Type 1 SF162 from Neutralization by Anti-gp120 and Anti-gp41 Antibodies. J. Virol. 2004, 78, 3279–3295. [Google Scholar] [CrossRef]
- Schellenbacher, C.; Roden, R.B.; Kirnbauer, R. Developments in L2-based human papillomavirus (HPV) vaccines. Virus Res. 2017, 231, 166–175. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, X.Y.; Frazer, I.H. Glycosylation of Human Papillomavirus Type 16 L1 Protein. Virology 1993, 194, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, R.; Zhang, L.; Shen, B.; Wang, N.; Xu, Q.; He, W.; He, W.; Li, G.; Su, S. Evolutionary changes of the novel Influenza D virus hemagglutinin-esterase fusion gene revealed by the codon usage pattern. Virulence 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Mutation-Selection Models of Codon Substitution and Their Use to Estimate Selective Strengths on Codon Usage. Mol. Biol. Evol. 2008, 25, 568–579. [Google Scholar] [CrossRef]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D. Selection on Codon Bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Chaney, J.L.; Clark, P. Roles for Synonymous Codon Usage in Protein Biogenesis. Annu. Rev. Biophys. 2015, 44, 143–166. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the Rate of Poliovirus Protein Synthesis through Large-Scale Codon Deoptimization Causes Attenuation of Viral Virulence by Lowering Specific Infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef]
- Cladel, N.M.; Bertotto, A.; Christensen, N.D. Human alpha and beta papillomaviruses use different synonymous codon profiles. Virus Genes 2010, 40, 329–340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, K.-N.; Liu, W.J.; Frazer, I. Codon usage bias and A+T content variation in human papillomavirus genomes. Virus Res. 2003, 98, 95–104. [Google Scholar] [CrossRef]
- Cid-Arregui, A.; Juárez, V.; Hausen, H.Z. A Synthetic E7 Gene of Human Papillomavirus Type 16 That Yields Enhanced Expression of the Protein in Mammalian Cells and Is Useful for DNA Immunization Studies. J. Virol. 2003, 77, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-T.; Tsai, Y.-C.; He, L.; Calizo, R.; Chou, H.-H.; Chang, T.-C.; Soong, Y.-K.; Hung, C.-F.; Lai, C.-H. A DNA Vaccine Encoding a Codon-Optimized Human Papillomavirus Type 16 E6 Gene Enhances CTL Response and Anti-tumor Activity. J. Biomed. Sci. 2006, 13, 481–488. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Sadeghnezhad, E. SADEG: Stability Analysis in Differentially Expressed Genes. 2017. Available online: https://CRAN.R-project.org/package=SADEG (accessed on 14 November 2020).
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- He, Z.; Gan, H.; Liang, X. Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef]
- Sharp, P.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Malik, Y.S.; Ansari, M.I.; Kattoor, J.J.; Kaushik, R.; Sircar, S.; Subbaiyan, A.; Tiwari, R.; Dhama, K.; Ghosh, S.; Tomar, S.; et al. Evolutionary and codon usage preference insights into spike glycoprotein of SARS-CoV-2. Brief Bioinform. 2020, 22, 1006–1022. [Google Scholar] [CrossRef]
- Qmichou, Z.; Khyatti, M.; Berraho, M.; Ennaji, M.M.; Benbacer, L.; Nejjari, C.; Benjaafar, N.; Benider, A.; Attaleb, M.; El Mzibri, M. Analysis of mutations in the E6 oncogene of human papillomavirus 16 in cervical cancer isolates from Moroccan women. BMC Infect. Dis. 2013, 13, 378. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yoshikawa, H.; Nakagawa, S.; Tang, X.; Yasugi, T.; Kawana, K.; Sekiya, S.; Hirai, Y.; Kukimoto, I.; Kanda, T.; et al. Enhanced oncogenicity of human papillomavirus type 16 (HPV16) variants in Japanese population. Cancer Lett. 2000, 156, 159–165. [Google Scholar] [CrossRef]
- Cai, H.; Chen, C.; Ding, X. Human papillomavirus type 16 E6 gene variations in Chinese population. Eur. J. Surg. Oncol. (EJSO) 2010, 36, 160–163. [Google Scholar] [CrossRef]
- Zhe, X.; Xin, H.; Pan, Z.; Jin, F.; Zheng, W.; Li, H.; Li, D.; Cao, D.; Li, Y.; Zhang, C.; et al. Genetic variations in E6, E7 and the long control region of human papillomavirus type 16 among patients with cervical lesions in Xinjiang, China. Cancer Cell Int. 2019, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, H.; Wu, K.; Shi, X.; Ma, S.; Sun, Q. Prevalence of HPV and variation of HPV 16/HPV 18 E6/E7 genes in cervical cancer in women in South West China. J. Med. Virol. 2014, 86, 1926–1936. [Google Scholar] [CrossRef]
- Shang, Q.; Wang, Y.; Fang, Y.; Wei, L.; Chen, S.; Sun, Y.; Li, B.; Zhang, F.; Gu, H. Human papillomavirus type 16 variant analysis of E6, E7, and L1 [corrected] genes and long control region in [corrected] cervical carcinomas in patients in northeast China. J. Clin. Microbiol. 2011, 49, 2656–2663. [Google Scholar] [CrossRef]
- Valle, G.F.; Banks, L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J. Gen. Virol. 1995, 76, 1239–1245. [Google Scholar] [CrossRef]
- Ellis, J.; Keating, P.; Baird, J.; Hounsell, E.F.; Renouf, D.V.; Rowe, M.; Hopkins, D.; Duggan-Keen, M.; Bartholomew, J.; Young, L.; et al. The association of an HPV16 oncogene variant with HLA-B7 has implications for vaccine design in cervical cancer. Nat. Med. 1995, 1, 464–470. [Google Scholar] [CrossRef]
- Vartanian, J.-P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for Editing of Human Papillomavirus DNA by APOBEC3 in Benign and Precancerous Lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Burns, M.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef]
- Wan, H.; Gao, J.; Yang, H.; Yang, S.; Harvey, R.; Chen, Y.Q.; Zheng, N.Y.; Chang, J.; Carney, P.J.; Li, X.; et al. The neuraminidase of A(H3N2) influenza viruses circulating since 2016 is antigenically distinct from the A/Hong Kong/4801/2014 vaccine strain. Nat. Microbiol. 2019, 4, 2216–2225. [Google Scholar] [CrossRef]
- Mondotte, J.A.; Lozach, P.-Y.; Amara, A.; Gamarnik, A.V. Essential Role of Dengue Virus Envelope Protein N Glycosylation at Asparagine-67 during Viral Propagation. J. Virol. 2007, 81, 7136–7148. [Google Scholar] [CrossRef]
- Godi, A.; Kemp, T.J.; Pinto, L.A.; Beddows, S. Sensitivity of Human Papillomavirus (HPV) Lineage and Sublineage Variant Pseudoviruses to Neutralization by Nonavalent Vaccine Antibodies. J. Infect. Dis. 2019, 220, 1940–1945. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, L.; He, W.; Zhang, X.; Wen, B.; Wang, C.; Xu, Q.; Li, G.; Zhou, J.; Veit, M.; et al. Genetic Evolution and Molecular Selection of the HE Gene of Influenza C Virus. Viruses 2019, 11, 167. [Google Scholar] [CrossRef]
- Hu, J.-S.; Wang, Q.-Q.; Zhang, J.; Chen, H.-T.; Xu, Z.-W.; Zhu, L.; Ding, Y.-Z.; Ma, L.-N.; Xu, K.; Gu, Y.-X.; et al. The characteristic of codon usage pattern and its evolution of hepatitis C virus. Infect. Genet. Evol. 2011, 11, 2098–2102. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF | Nucleotide Mutation | Amino Acid Mutation | Proportion of Sequences with the Corresponding Mutations in Each Sublineage (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | B1 | C1 | D1 | D2 | D3 | D4 | |||
| (n = 1053) | (n = 204) | (n = 11) | (n = 84) | (n = 28) (B, n = 34) | (n = 50) (C, n = 56) | (n = 12) | (n = 35) | (n = 95) | (n = 12) | |||
| E1 | T1220C | V119A | 100 | 1.1 | ||||||||
| C1415T | T184I | 100 (89) | ||||||||||
| C1598T | P245L | 92.9 (76) | 1.1 | |||||||||
| A1667G | H268R | 100 | 100 | 100 | 100 | |||||||
| T2252C | F463S | 0.1 | 91.7 | |||||||||
| T2253C | F463S | 91.7 | ||||||||||
| T2342C | F493S | 0.1 | 1.2 | 100 | 95.8 | |||||||
| C2343T | F493S | 100 | 94.7 | |||||||||
| T2354C | L497P | 0.5 | 100 (100) | |||||||||
| T2375C | L504P | 0.3 | 2.5 | 3.6 (2.9) | 100 | |||||||
| C2456T | T531I | 0.1 | 99.0 | |||||||||
| E2 | C3158G | T135R | 100 | |||||||||
| A3180C | E142D | 14.3 (14.7) | 6 (7.1) | 97.9 | ||||||||
| T3223A | L157I/M a | 100 | 100 | 100 | 100 | |||||||
| T3383C | I210T | 1.7 | 100 | 100 | ||||||||
| T3386C | I211T | 95.8 | ||||||||||
| G3412A | A220T | 100 | ||||||||||
| G3415A | A221T | 100 | ||||||||||
| G3430A | A226T | 100 (98.2) | 2.9 | |||||||||
| E5 | G3881A | A7T | 100 (100) | |||||||||
| A4054T | L64S/F b | 100 (100) | 2 (1.8) | |||||||||
| A4089T | H76L | 97.6 | ||||||||||
| E6 | G132T | R10I | 98 (87.5) | |||||||||
| C143G | Q14D | 92.9 (98.2) | 98 (99.5) | |||||||||
| T350G | L83V | 47.8 | 21.6 | 3.6 (14.7) | 100 | 100 | 100 | 100 | ||||
| E7 | A647G | N29S | 98.8 | 100 (89.3) | ||||||||
| L1 | A6178C | N207T | 41.7 | 14.3 (11.8) | 78 (75) | 8.3 | 5.7 | 100 | ||||
| T6480C | S308P | 3.6 (2.9) | 100 (100) | |||||||||
| A6801T | T415S | 97.9 | ||||||||||
| L2 | A4967G | T245A | 0.1 | 100 | 97.1 | 98.9 | 100 | |||||
| A5032T | L266F | 100 | 100 | 100 | 100 | |||||||
| A5288C | T353P | 100 (89.3) | ||||||||||
| A5288G | T353A | 100 (97.1) | ||||||||||
| T5366G | S379A/V c | 100 | 97.1 | 96.8 | 100 | |||||||
| T5384G | S385A | 100 | 97.1 | 100 | 100 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Li, J.; Du, H.; Ou, Z. Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16. Viruses 2021, 13, 1281. https://doi.org/10.3390/v13071281
Liu W, Li J, Du H, Ou Z. Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16. Viruses. 2021; 13(7):1281. https://doi.org/10.3390/v13071281
Chicago/Turabian StyleLiu, Wei, Junhua Li, Hongli Du, and Zhihua Ou. 2021. "Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16" Viruses 13, no. 7: 1281. https://doi.org/10.3390/v13071281
APA StyleLiu, W., Li, J., Du, H., & Ou, Z. (2021). Mutation Profiles, Glycosylation Site Distribution and Codon Usage Bias of Human Papillomavirus Type 16. Viruses, 13(7), 1281. https://doi.org/10.3390/v13071281