
Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy


Abstract
1. Adeno-Associated Virus: Discovery and Biology
2. Overview of Cancer Gene Therapy Approaches with rAAV
3. WT AAV Induces Cancer Cell Death
4. Combination Therapy—rAAV and Chemotherapy
5. Targeting Cancer Stem Cells with AAV
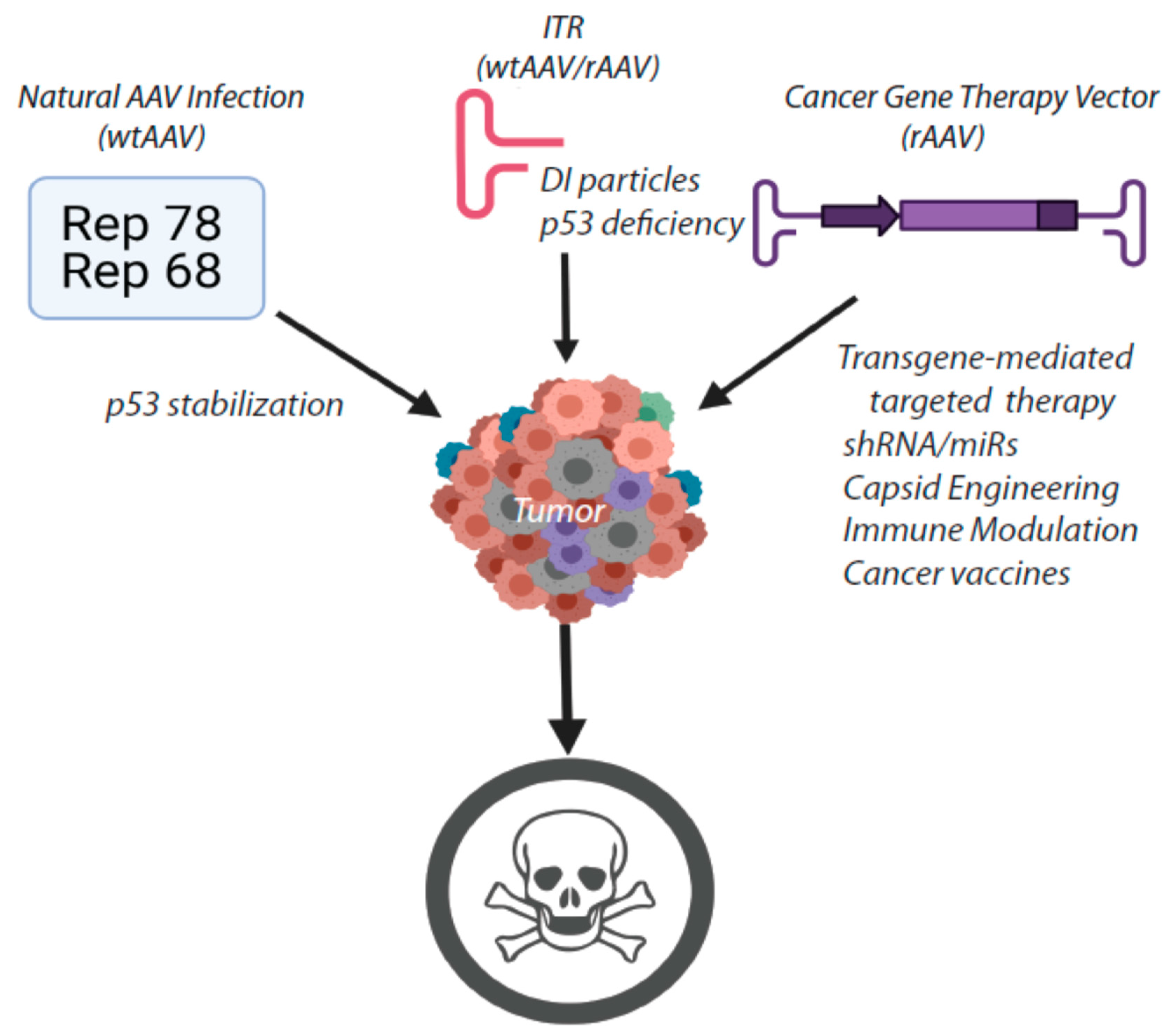
6. Closing Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Earley, L.F.; Conatser, M.L.; Lue, V.; Dobbins, M.A.L.; Li, C.; Hirsch, M.L.; Samulski, R.J. Adeno-Associated Virus Serotype-Specific Inverted Terminal Repeat Sequence Role in Vector Transgene Expression. Hum. Gene Ther. 2020, 31, 151–162. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-Associated Virus Type 2 Capsids with Externalized VP1/VP2 Trafficking Domains Are Generated prior to Passage through the Cytoplasm and Are Maintained until Uncoating Occurs in the Nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef]
- Cao, M.; You, H.; Hermonat, P.L. The X Gene of Adeno-Associated Virus 2 (AAV2) Is Involved in Viral DNA Replication. PLoS ONE 2014, 9, e104596. [Google Scholar] [CrossRef]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef]
- Hermonat, P.L.; Santin, A.D.; De Greve, J.; De Rijcke, M.; Bishop, B.M.; Han, L.; Mane, M.; Kokorina, N. Chromosomal latency and expression at map unit 96 of a wild-type plus adeno-associated virus (AAV)/Neo vector and identification of p81, a new AAV transcriptional promoter. J. Hum. Virol. 2000, 2, 359–368. [Google Scholar]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Choi, V.W.; Samulski, R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006, 1, 1412–1428. [Google Scholar] [CrossRef]
- Buller, R.M.L.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef]
- Im, D.-S.; Muzyczka, N. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell 1990, 61, 447–457. [Google Scholar] [CrossRef]
- Murphy, M.; Gomos-Klein, J.; Stankic, M.; Falck-Pedersen, E. Adeno-Associated Virus Type 2 p5 Promoter: A Rep-Regulated DNA Switch Element Functioning in Transcription, Replication, and Site-Specific Integration. J. Virol. 2007, 81, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Kyöstiö, S.R.; Wonderling, R.S.; Owens, R.A. Negative regulation of the adeno-associated virus (AAV) P5 promoter involves both the P5 rep binding site and the consensus ATP-binding motif of the AAV Rep68 protein. J. Virol. 1995, 69, 6787–6796. [Google Scholar] [CrossRef]
- Kokorina, N.A.; Santin, A.D.; Li, C.; Hermonat, P.L. Involvement of protein-DNA interaction in adeno-associated virus Rep78-mediated inhibition of HIV-1. J. Hum. Virol. 1999, 1, 441–450. [Google Scholar]
- Hermonat, P.L. Inhibition of H-ras expression by the adeno-associated virus Rep78 transformation suppressor gene product. Cancer Res. 1991, 51, 3373–3377. [Google Scholar]
- Hermonat, P. Down-regulation of the human c-fos and c-myc proto-oncogene promoters by adeno-associated virus Rep78. Cancer Lett. 1994, 81, 129–136. [Google Scholar] [CrossRef]
- Wonderling, R.S.; Owens, R.A. Binding sites for adeno-associated virus Rep proteins within the human genome. J. Virol. 1997, 71, 2528–2534. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious Entry Pathway of Adeno-Associated Virus and Adeno-Associated Virus Vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Gao, G.P.; Weitzman, M.D.; DeMatteo, R.; Burda, J.F.; Wilson, J.M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996, 70, 520–532. [Google Scholar] [CrossRef]
- Boucher, D.W.; Melnick, J.L.; Mayor, H.D. Nonencapsidated Infectious DNA of Adeno-Satellite Virus in Cells Coinfected with Herpesvirus. Science 1971, 173, 1243–1245. [Google Scholar] [CrossRef]
- Meyers, C.; Alam, S.; Mane, M.; Hermonat, P.L. Altered Biology of Adeno-associated Virus Type 2 and Human Papillomavirus during Dual Infection of Natural Host Tissue. Virology 2001, 287, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, S.C.; Li, C.; Hirsch, M.L.; Setola, V.; Samulski, R.J. Identification and Validation of Small Molecules That Enhance Recombinant Adeno-associated Virus Transduction following High-Throughput Screens. J. Virol. 2016, 90, 7019–7031. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Bai, Y.; Shao, Y.; Bai, J.; Ma, Z.; Liu, Q.; Wu, S. Recombinant adeno-associated virus expressing a p53-derived apoptotic peptide (37AA) inhibits HCC cells growth in vitro and in vivo. Oncotarget 2017, 8, 16801–16810. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qazilbash, M.; Xiao, X.; Seth, P.; Cowan, K.; Walsh, C. Cancer gene therapy using a novel adeno-associated virus vector expressing human wild-type p53. Gene Ther. 1997, 4, 675–682. [Google Scholar] [CrossRef]
- Ng, S.S.M.; Gao, Y.; Chau, D.H.W.; Li, G.H.Y.; Lai, L.H.; Huang, P.T.; Huang, C.F.; Huang, J.J.; Chen, Y.; Kung, H.F.; et al. A novel glioblastoma cancer gene therapy using AAV-mediated long-term expression of human TERT C-terminal polypeptide. Cancer Gene Ther. 2007, 14, 561–572. [Google Scholar] [CrossRef]
- Noro, T.; Miyake, K.; Suzuki-Miyake, N.; Igarashi, T.; Uchida, E.; Misawa, T.; Yamazaki, Y.; Shimada, T. Adeno-Associated Viral Vector-Mediated Expression of Endostatin Inhibits Tumor Growth and Metastasis in an Orthotropic Pancreatic Cancer Model in Hamsters. Cancer Res. 2004, 64, 7486–7490. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.S.; Chang, B.; Lin, J.; Case, S.S.; Luan, B.; Wu-Prior, W.-W.; VanRoey, M.; Jooss, K. Anti-Tumor Efficacy of Human Angiostatin Using Liver-Mediated Adeno-Associated Virus Gene Therapy. Mol. Ther. 2004, 9, 56–66. [Google Scholar] [CrossRef]
- Zi-Bo, L.I.; Zhao-Jun, Z.E.N.G.; Qian, C.H.E.N.; Sai-Qun, L.U.O.; Wei-Xin, H.U. Recombinant AAV-mediated HSVtk gene transfer with direct intratumoral injections and Tet-On regulation for implanted human breast cancer. BMC Cancer 2006, 6, 66. [Google Scholar] [CrossRef]
- Zeng, Z.-J.; Xiang, S.-G.; Xue, W.-W.; Li, H.-D.; Ma, N.; Ren, Z.-J.; Xu, Z.-J.; Jiao, C.-H.; Wang, C.-Y.; Hu, W.-X. The cell death and DNA damages caused by the Tet-On regulating HSV-tk/GCV suicide gene system in MCF-7 cells. Biomed. Pharmacother. 2014, 68, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hicks, M.; Kaminsky, S.; Moore, M.A.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The Triple Negative Paradox: Primary Tumor Chemosensitivity of Breast Cancer Subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, Z.; Zhang, D.; Zhang, W.; Yan, Q.; Shi, X.; Zhang, M.; Zhao, Y.; Zhang, Y.; Jiang, B.; et al. A novel colon cancer gene therapy using rAAV-mediated expression of human shRNA-FHL2. Int. J. Oncol. 2013, 43, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Saga, Y.; Uchibori, R.; Tsukahara, T.; Urabe, M.; Kume, A.; Fujiwara, H.; Suzuki, M.; Ozawa, K.; Mizukami, H. Eradication of cervical cancer in vivo by an AAV vector that encodes shRNA targeting human papillomavirus type 16 E6/E7. Int. J. Oncol. 2018, 52, 687–696. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, S.; Collins, S.; Van Pijkeren, J.P.; O’Hanlon, D.; O’Sullivan, G.C.; Tangney, M. Targeting of breast metastases using a viral gene vector with tumour-selective transcription. Anticancer. Res. 2011, 31, 1627–1635. [Google Scholar]
- Münch, R.C.; Janicki, H.; Völker, I.; Rasbach, A.; Hallek, M.; Büning, H.; Buchholz, C.J. Displaying High-affinity Ligands on Adeno-associated Viral Vectors Enables Tumor Cell-specific and Safe Gene Transfer. Mol. Ther. 2013, 21, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.; Ortiz, L.; Ling, C.; Rivers, A.E.; Aslanidi, G. Rationally designed capsid and transgene cassette of AAV6 vectors for dendritic cell-based cancer immunotherapy. Immunol. Cell Biol. 2013, 92, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Judd, J.; Ho, M.L.; Tiwari, A.; Gomez, E.J.; Dempsey, C.; Van Vliet, K.; Igoshin, O.A.; Silberg, J.J.; Agbandje-McKenna, M.; Suh, J. Tunable Protease-Activatable Virus Nanonodes. ACS Nano 2014, 8, 4740–4746. [Google Scholar] [CrossRef]
- Maguire, C.A.; Gianni, D.; Meijer, D.H.; Shaket, L.A.; Wakimoto, H.; Rabkin, S.D.; Gao, G.; Sena-Esteves, M. Directed evolution of adeno-associated virus for glioma cell transduction. J. Neuro Oncol. 2010, 96, 337–347. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Y.; Liu, S.; Kung, H.F.; Sun, X.; Zheng, D.; Xu, R. Recombinant adeno-associated virus-mediated TRAIL gene therapy suppresses liver metastatic tumors. Int. J. Cancer 2005, 116, 314–321. [Google Scholar] [CrossRef]
- Hensel, J.A.; Khattar, V.; Ashton, R.; Ponnazhagan, S. Recombinant AAV-CEA Tumor Vaccine in Combination with an Immune Adjuvant Breaks Tolerance and Provides Protective Immunity. Mol. Ther. Oncolytics 2019, 12, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cukor, G.; Blacklow, N.R.; Kibrick, S.; Swan, I.C.; Sidney, K. Effect of Adeno-Associated Virus on Cancer Expression by Herpesvirus-Transformed Hamster Cells 2. J. Natl. Cancer Inst. 1975, 55, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Hermonat, P.L. The adeno-Associated virus Rep78 gene inhibits cellular transformation induced by bovine papillomavirus. Virology 1989, 172, 253–261. [Google Scholar] [CrossRef]
- Hermonat, P.L. Adeno-associated virus inhibits human papillomavirus type 16: A viral interaction implicated in cervical cancer. Cancer Res. 1994, 54, 2278–2281. [Google Scholar] [PubMed]
- Hermonat, P.L. Inhibition of bovine papillomavirus plasmid DNA replication by adeno-associated virus. Virology 1992, 189, 329–333. [Google Scholar] [CrossRef]
- De La Maza, L.M.; Carter, B.J. Inhibition of adenovirus oncogenicity in hamsters by adeno-associated virus DNA. J. Natl. Cancer Inst. 1981, 67, 1323–1326. [Google Scholar]
- Khleif, S.N.; Myersz, T.; Carter, B.J.; Trempe, J.P. Inhibition of Cellular transformation by the adeno-associated virus rep gene. Virology 1991, 181, 738–741. [Google Scholar] [CrossRef]
- Batchu, R.B.; Shammas, M.A.; Wang, J.Y.; Munshi, N.C. Interaction of adeno-associated virus Rep78 with p53: Implications in growth inhibition. Cancer Res. 1999, 59, 3592–3595. [Google Scholar]
- Alam, S.; Meyers, C. Adeno-associated virus type 2 induces apoptosis in human papillomavirus-infected cell lines but not in normal keratinocytes. J. Virol. 2009, 83, 10286–91022. [Google Scholar] [CrossRef][Green Version]
- Alam, S.; Bowser, B.S.; Conway, M.J.; Israr, M.; Tandon, A.; Meyers, C. Adeno-associated virus type 2 infection activates caspase dependent and independent apoptosis in multiple breast cancer lines but not in normal mammary epithelial cells. Mol. Cancer 2011, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Bowser, B.S.; Israr, M.; Conway, M.J.; Meyers, C. Adeno-associated virus type 2 infection of nude mouse human breast cancer xenograft induces necrotic death and inhibits tumor growth. Cancer Biol. Ther. 2014, 15, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Afione, S.; Kotin, R.M. Adeno-Associated Virus Type 2 Rep78 Induces Apoptosis through Caspase Activation Independently of p53. J. Virol. 2000, 74, 9441–9450. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Raj, K.; Saudan, P.; Beard, P. How adeno-associated virus Rep78 protein arrests cells completely in S phase. Proc. Natl. Acad. Sci. USA 2005, 102, 13634–13639. [Google Scholar] [CrossRef]
- Saudan, P.; Vlach, J.; Beard, P. Inhibition of S-phase progression by adeno-associated virus Rep78 protein is mediated by hypophosphorylated pRb. EMBO J. 2000, 19, 4351–4361. [Google Scholar] [CrossRef]
- Russell, D.W.; Miller, A.D.; Alexander, I.E. Adeno-associated virus vectors preferentially transduce cells in S phase. Proc. Natl. Acad. Sci. USA 1994, 91, 8915–8919. [Google Scholar] [CrossRef]
- Raj, K.; Ogston, P.; Beard, P. Virus-mediated killing of cells that lack p53 activity. Nat. Cell Biol. 2001, 412, 914–917. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Fagan, B.M.; Dumitru, R.; Bower, J.J.; Yadav, S.; Porteus, M.H.; Pevny, L.H.; Samulski, R.J. Viral Single-Strand DNA Induces p53-Dependent Apoptosis in Human Embryonic Stem Cells. PLoS ONE 2011, 6, e27520. [Google Scholar] [CrossRef]
- Coker, A.L.; Russell, R.B.; Bond, S.M.; Pirisi, L.; Liu, Y.; Mane, M.; Kokorina, N.; Gerasimova, T.; Hermonat, P.L. Adeno-Associated Virus Is Associated with a Lower Risk of High-Grade Cervical Neoplasia. Exp. Mol. Pathol. 2001, 70, 83–89. [Google Scholar] [CrossRef]
- Mayor, H.D.; Drake, S.; Stahmann, J.; Mumford, D.M. Antibodies to adeno-associated satellite virus and herpes simplex in sera from cancer patients and normal adults. Am. J. Obstet. Gynecol. 1976, 126, 100–104. [Google Scholar] [CrossRef]
- Georg-Fries, B.; Biederlack, S.; Wolf, J.; Hausen, H.Z. Analysis of proteins, helper dependence, and seroepidemiology of a new human parvovirus. Virology 1984, 134, 64–71. [Google Scholar] [CrossRef]
- Han, L.; Parmley, T.H.; Keith, S.; Kozlowski, K.J.; Smith, L.J.; Hermonat, P.L. High prevalence of adeno-associated virus (AAV) type 2 rep DNA in cervical materials: AAV may be sexually transmitted. Virus Genes 1996, 12, 47–52. [Google Scholar] [CrossRef]
- Tobiasch, E.; Rabreau, M.; Geletneky, K.; Laruë-Charlus, S.; Severin, F.; Becker, N.; Schlehofer, J.R. Detection of adeno-associated virus DNA in human genital tissue and in material from spontaneous abortion. J. Med Virol. 1994, 44, 215–222. [Google Scholar] [CrossRef]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Qin, W.; Xu, G.; Tai, P.W.L.; Wang, C.; Luo, L.; Li, C.; Hu, X.; Xue, J.; Lu, Y.; Zhou, Q.; et al. Large-scale molecular epidemiological analysis of AAV in a cancer patient population. Oncogene 2021, 40, 3060–3071. [Google Scholar] [CrossRef]
- Zacharias, J.; Romanova, L.G.; Menk, J.; Philpott, N.J. p53 inhibits adeno-associated viral vector integration. Hum. Gene. Ther. 2011, 22, 1445–1451. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar]
- Chien, J.; Kuang, R.; Landen, C.; Shridhar, V. Platinum-Sensitive Recurrence in Ovarian Cancer: The Role of Tumor Microenvironment. Front. Oncol. 2013, 3, 251. [Google Scholar] [CrossRef]
- Bastola, P.; Neums, L.; Schoenen, F.; Chien, J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol. Oncol. 2016, 10, 1559–1574. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Subramanian, I.V. Adeno-Associated Virus-Mediated Delivery of a Mutant Endostatin in Combination with Carboplatin Treatment Inhibits Orthotopic Growth of Ovarian Cancer and Improves Long-term Survival. Cancer Res. 2006, 66, 4319–4328. [Google Scholar] [CrossRef] [PubMed]
- Dang, S.C.; Feng, S.; Wang, P.J.; Cui, L.; Qu, J.G.; Zhang, J.X. Overexpression of Survivin mutant Thr34Ala induces apoptosis and inhibits gastric cancer growth. Neoplasma 2015, 62, 81–87. [Google Scholar] [CrossRef][Green Version]
- Tu, S.P.; Xue, Z.; Sun, P.H.; Zhu, L.M.; Jiang, S.H.; Qiao, M.M.; Chi, A.L. Adeno-associated virus-mediated survivin mutant Thr34Ala cooperates with oxaliplatin to inhibit tumor growth and angiogenesis in colon cancer. Oncol. Rep. 2011, 25, 1039–1046. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Huang, F.; Cai, H.; Wu, Y.; He, G.; Tan, W.-S. The efficacy of combination therapy using adeno-associated virus-TRAIL targeting to telomerase activity and cisplatin in a mice model of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1827–1837. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, Z.; Xiang, Y.; Ma, H.; Liu, S.; Liu, Y.; Zheng, D. Synergistic antitumor effect of AAV-mediated TRAIL expression combined with cisplatin on head and neck squamous cell carcinoma. BMC Cancer 2011, 11, 54. [Google Scholar] [CrossRef]
- Yan, Z.; Zak, R.; Zhang, Y.; Ding, W.; Godwin, S.; Munson, K.; Peluso, R.; Engelhardt, J.F. Distinct Classes of Proteasome-Modulating Agents Cooperatively Augment Recombinant Adeno-Associated Virus Type 2 and Type 5-Mediated Transduction from the Apical Surfaces of Human Airway Epithelia. J. Virol. 2004, 78, 2863–2874. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Cochrane, M.; McIntosh, J.; Ng, C.Y.; Zhou, J.; Gray, J.T.; Davidoff, A.M. Enhancing transduction of the liver by adeno-associated viral vectors. Gene Ther. 2008, 16, 60–69. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Li, C.; Samulski, R.J. Arsenic Trioxide Stabilizes Accumulations of Adeno-Associated Virus Virions at the Perinuclear Region, Increasing Transduction In Vitro and In Vivo. J. Virol. 2013, 87, 4571–4583. [Google Scholar] [CrossRef]
- Zhong, L.; Qing, K.; Si, Y.; Chen, L.; Tan, M.; Srivastava, A. Heat-shock Treatment-mediated Increase in Transduction by Recombinant Adeno-associated Virus 2 Vectors Is Independent of the Cellular Heat-shock Protein 90. J. Biol. Chem. 2004, 279, 12714–12723. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Song, L.; Tao, X.; Lin, L.; Chen, C.; Yao, H.; He, G.; Zou, G.; Cao, Z.; Yan, S.; Lu, L.; et al. Cerasomal Lovastatin Nanohybrids for Efficient Inhibition of Triple-Negative Breast Cancer Stem Cells To Improve Therapeutic Efficacy. ACS Appl. Mater. Interfaces 2018, 10, 7022–7030. [Google Scholar] [CrossRef]
- Carvalho, J. Cell Reversal from a Differentiated to a Stem-Like State at Cancer Initiation. Front. Oncol. 2020, 10, 541. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Hoque, M.O. Targeting Cancer Stem Cells: A Strategy for Effective Eradication of Cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef]
- Cross, D.; Burmester, J.K. Gene Therapy for Cancer Treatment: Past, Present and Future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar] [CrossRef]
- Ajith, T.A. Strategies used in the clinical trials of gene therapy for cancer. J. Exp. Ther. Oncol. 2015, 11, 33–39. [Google Scholar]
- Brown, N.; Song, L.; Kollu, N.R.; Hirsch, M.L. Adeno-Associated Virus Vectors and Stem Cells: Friends or Foes? Hum. Gene Ther. 2017, 28, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.J.; Ul-Hasan, T.; Carvaines, S.K.; Van Vliet, K.; Yang, E.; Wong, K.K.; Agbandje-McKenna, M.; Chatterjee, S. Gene Transfer Properties and Structural Modeling of Human Stem Cell-derived AAV. Mol. Ther. 2014, 22, 1625–1634. [Google Scholar] [CrossRef]
- Song, L.; Kauss, M.A.; Kopin, E.; Chandra, M.; Ul-Hasan, T.; Miller, E.; Jayandharan, G.R.; Rivers, A.E.; Aslanidi, G.V.; Ling, C.; et al. Optimizing the transduction efficiency of capsid-modified AAV6 serotype vectors in primary human hematopoietic stem cells in vitro and in a xenograft mouse model in vivo. Cytotherapy 2013, 15, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Song, Z.; Fry, N.J.; Conatser, L.; Llanga, T.; Mei, H.; Kafri, T.; Hirsch, M.L. Gene Delivery to Human Limbal Stem Cells Using Viral Vectors. Hum. Gene Ther. 2019, 30, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.T.; Parylak, S.L.; Kim, S.; Mac, N.; Lim, C.K.; Gallina, I.S.; Bloyd, C.W.; Newberry, A.; Saavedra, C.D.; Novák, O.; et al. AAV Ablates Neurogenesis in the Adult Murine Hippocampus. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rapti, K.; Stillitano, F.; Karakikes, I.; Nonnenmacher, M.; Weber, T.; Hulot, J.-S.; Hajjar, R.J. Effectiveness of gene delivery systems for pluripotent and differentiated cells. Mol. Ther. Methods Clin. Dev. 2015, 2, 14067. [Google Scholar] [CrossRef] [PubMed]
- Kirschen, G.W.; Kery, R.; Liu, H.; Ahamad, A.; Chen, L.; Akmentin, W.; Kumar, R.; Levine, J.; Xiong, Q.; Ge, S. Genetic dissection of the neuro-glio-vascular machinery in the adult brain. Mol. Brain 2018, 11, 2. [Google Scholar] [CrossRef]
- Yang, H.; Qing, K.; Keeler, G.D.; Yin, L.; Mietzsch, M.; Ling, C.; Hoffman, B.E.; Agbandje-McKenna, M.; Tan, M.; Wang, W.; et al. Enhanced Transduction of Human Hematopoietic Stem Cells by AAV6 Vectors: Implications in Gene Therapy and Genome Editing. Mol. Ther. Nucleic Acids 2020, 20, 451–458. [Google Scholar] [CrossRef]
- Cromer, M.K.; Camarena, J.; Martin, R.M.; Lesch, B.J.; Vakulskas, C.A.; Bode, N.M.; Kurgan, G.; Collingwood, M.A.; Rettig, G.R.; Behlke, M.A.; et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021, 27, 677–687. [Google Scholar] [CrossRef]
- Cromer, M.K.; Vaidyanathan, S.; Ryan, D.E.; Curry, B.; Lucas, A.B.; Camarena, J.; Kaushik, M.; Hay, S.; Martin, R.M.; Steinfeld, I.; et al. Global Transcriptional Response to CRISPR/Cas9-AAV6-Based Genome Editing in CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. 2018, 26, 2431–2442. [Google Scholar] [CrossRef]
- Basche, M.; Kampik, D.; Kawasaki, S.; Branch, M.J.; Robinson, M.; Larkin, D.F.; Smith, A.J.; Ali, R.R.; Larkin, F. Sustained and Widespread Gene Delivery to the Corneal Epithelium via In Situ Transduction of Limbal Epithelial Stem Cells, Using Lentiviral and Adeno-Associated Viral Vectors. Hum. Gene Ther. 2018, 29, 1140–1152. [Google Scholar] [CrossRef]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Verdera, H.C.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J., Jr.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13, eabd3438. [Google Scholar] [CrossRef]
- Espín-Palazón, R.; Stachura, D.L.; Campbell, C.A.; García-Moreno, D.; Del Cid, N.; Kim, A.D.; Candel, S.; Meseguer, J.; Mulero, V.; Traver, D. Proinflammatory Signaling Regulates Hematopoietic Stem Cell Emergence. Cell 2014, 159, 1070–1085. [Google Scholar] [CrossRef]
- Berns, K.I. The Unusual Properties of the AAV Inverted Terminal Repeat. Hum. Gene Ther. 2020, 31, 518–523. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Journal of Gene Medicine Database Trial ID | Start Date | Disease | Payload | Phase |
|---|---|---|---|---|
| CH-0025 | 2001 | Malignant Melanoma | GM-CSF B7.2 | 1 |
| CN-0020 | 2008 | Malignant Solid Tumors | Tumor Antigen | 1 |
| CN-0028 | 2012 | Gastric Cancer | Carcinoembryonic antigen (CEA) | 1 |
| ES-0021 | 2012 | Pancreatic Cancer | Hyaluronidase | 1 |
| JP-0014 | N/A | Hormone refractory metastatic prostate cancer | HSV-TK | 1 |
| NL-0012 | 2004 | Hormone refractory prostate cancer | GM-CSF | 1 |
| NL-0013 | 2006 | Metastatic Prostate Cancer | GM-CSF | 3 |
| NL-0014 | 2006 | Prostate Cancer | GM-CSF | 3 |
| NL-0015 | 2006 | Prostate Cancer | GM-CSF | 3 |
| NL-0016 | N/A | Prostate Cancer | GM-CSF | 1 |
| NL-0021 | 2005 | Prostate Cancer | IL-12 | 1 |
| UK-0133 | 2005 | Prostate Cancer | GM-CSF | 3 |
| UK-0134 | 2005 | Prostate Cancer | GM-CSF | 3 |
| US-0459 | 2001 | Hormone-Refractory Prostate Cancer | GM-CSF | 1 |
| US-0493 | 2001 | Hormone Refractory Prostate Cancer | GM-CSF | 1/2 |
| US-0653 | 2004 | Hormone-Refractory Prostate Cancer | GM-CSF | 3 |
| US-0675 | 2004 | Prostate Cancer | GM-CSF | 1/2 |
| US-0708 | 2005 | Prostate Cancer | GM-CSF | 3 |
| US-0903 | 2008 | Prostate Cancer | GM-CSF | 2 |
| US-1165 | 2012 | Prostate Cancer | GM-CSF | 1/2 |
| US-1748 | 2018 | Non-Hodgkin’s Lymphoma/B-cell Acute Lymphoblastic Leukemia | CD19, CD8a, N6 and TCRζ | 1 |
| US-1800 | 2018 | Multiple Myeloma | CAR2-α-BCMA, CD28/CD3ζ | 1 |
| Cancer Type | Chemotherapy | Transgene | rAAV Capsid, Promoter | References |
|---|---|---|---|---|
| Ovarian | Carboplatin | Endostatin (P125A) | Unknown, CGA | [75] |
| Ovarian | Topotecan and Paclitaxel | Bevacizumab Ab | rh.10, CGA | [31] |
| Gastric | 5-FU | Survivin (T34A) | Unknown, CGA | [76] |
| Colorectal | Oxaliplatin | Survivin (T34A) | Unknown, CGA | [77] |
| Colorectal | 5-FU | shRNA FHL2 | AAV2, U6 | [34] |
| Hepatocellular | Cisplatin | TRAIL | AAV2, hTERT | [78] |
| Head and Neck | Cisplatin | TRAIL | Unknown, CGA | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bower, J.J.; Song, L.; Bastola, P.; Hirsch, M.L. Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. Viruses 2021, 13, 1205. https://doi.org/10.3390/v13071205
Bower JJ, Song L, Bastola P, Hirsch ML. Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. Viruses. 2021; 13(7):1205. https://doi.org/10.3390/v13071205
Chicago/Turabian StyleBower, Jacquelyn J., Liujiang Song, Prabhakar Bastola, and Matthew L. Hirsch. 2021. "Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy" Viruses 13, no. 7: 1205. https://doi.org/10.3390/v13071205
APA StyleBower, J. J., Song, L., Bastola, P., & Hirsch, M. L. (2021). Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. Viruses, 13(7), 1205. https://doi.org/10.3390/v13071205