
The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence

 ,
,  , , , ,
, , , ,
Abstract
1. Introduction
2. Alphaviruses
Lifecycle of Alphavirus
3. The Functions of Alphavirus nsP3
3.1. Replication
3.2. Vector Specificity Determinants
3.3. Viral Virulence Determinants
3.4. Regulation of Host Stress Responses
3.5. Transmission Agent for Inter-Host and among Hosts
4. The Protein Domains of Alphavirus nsP3
4.1. Macrodomain
4.2. Alphavirus Unique Domain (AUD)
4.3. Hypervariable Domain
5. The Functions of Alphavirus nsP3 HVD
5.1. Alphavirus Proline-Rich Region Interactions with SH3-Domain of Host Cellular Proteins
5.2. Alphavirus FGDF-Like Motif Interactions with Host Cellular G3BP and Rasputin
5.3. Alphavirus nsP3 HVD Interactions with Other Host Cellular Proteins
5.4. Opal Stop Codon
6. Indel, Repetition and Duplication Events of Alphavirus nsP3 HVD
6.1. Indel, Repetition, and Duplication Events in CHIKV nsP3
6.2. Indel, Repetition and Duplication Events in AURAV nsP3
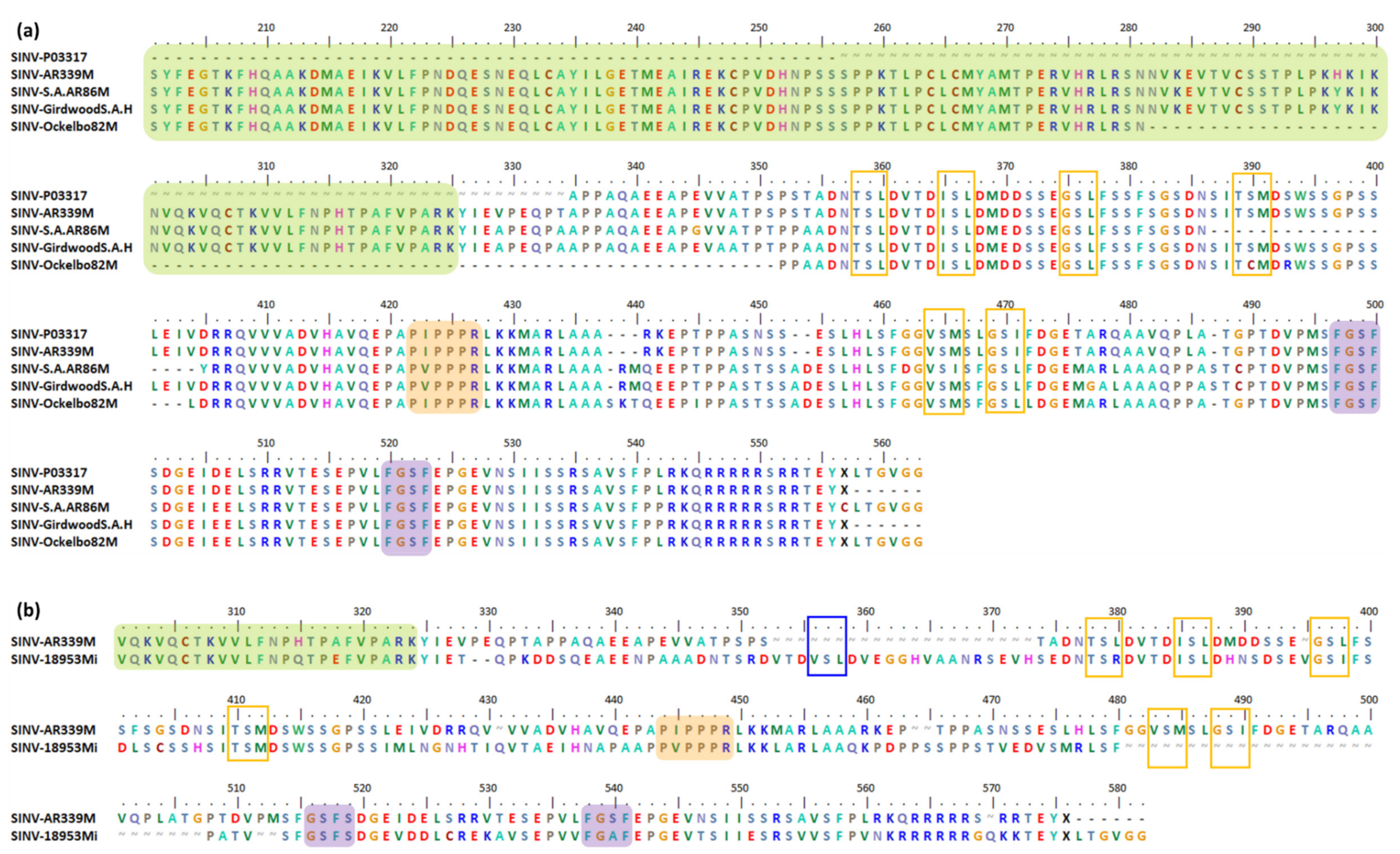
6.3. Indel, Repetition and Duplication Events in SINV nsP3
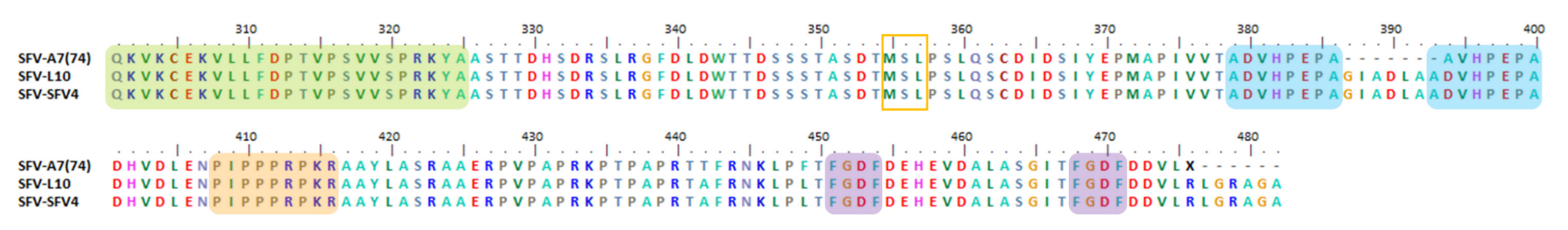
6.4. Indel, Repetition and Duplication Events in SFV nsP3
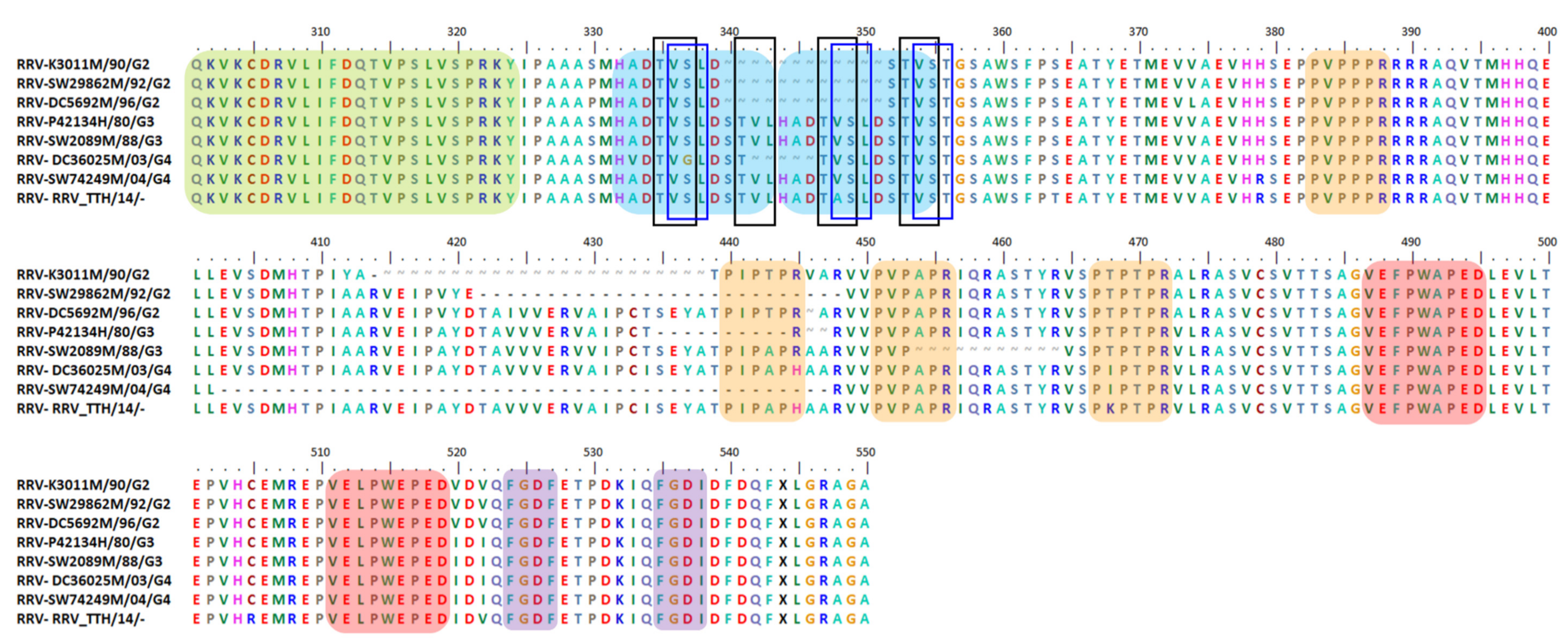
6.5. Indel, Repetition and Duplication Events in RRV nsP3 HVD
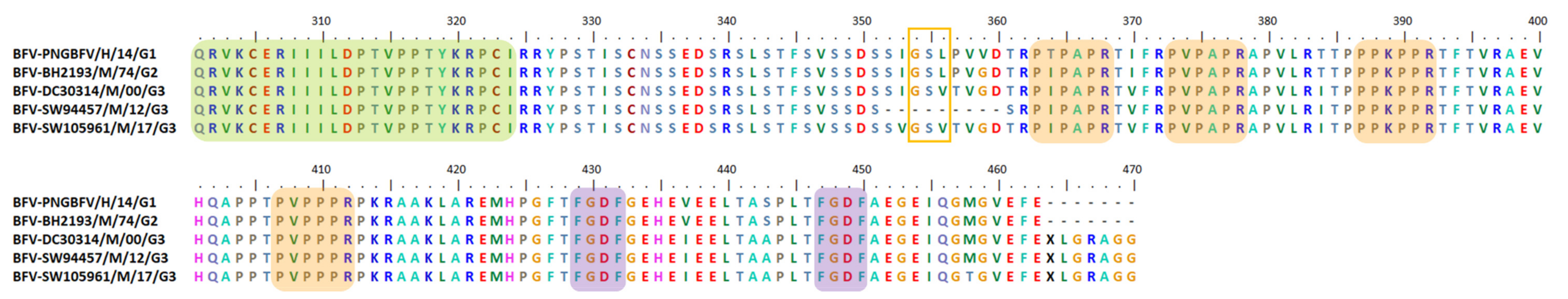
6.6. Indel, Repetition and Duplication Events in BFV nsP3 HVD
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virus Pathogen Resource (ViPR). Togaviridae. 2020. Available online: https://www.viprbrc.org/brc/home.spg?decorator=toga (accessed on 3 January 2020).
- Meshram, D.C.; Agback, P.; Shiliaev, N.; Urakova, N.; Mobley, J.A.; Agback, T.; Frolova, E.I.; Frolov, I. Multiple host factors interact with hypervariable domain of Chikungunya virus nsP3 and determine viral replication in cell-specific mode. J. Virol. 2018, 92, e00838-18. [Google Scholar] [CrossRef]
- Gotte, B.; Liu, L.; McInerney, G.M. The enigmatic Alphavirus non-structural protein 3 (nsP3) revealing its secrets at last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Earnest, J.T.; Kim, A.S.; Winkler, E.S.; Desai, P.; Adams, L.J.; Hu, G.; Bullock, C.; Gold, B.; Cherry, S.; et al. Expression of the Mxra8 receptor promotes alphavirus infection and pathogenesis in mice and drosophila. Cell Rep. 2019, 28, e5. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.A.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary relationships and systematics of the alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef]
- Kääriäinen, L.; Ahola, T. Functions of alphavirus nonstructural proteins in RNA replication. Prog. Nucleic Acid Res. Mol. Biol. 2002, 71, 187–222. [Google Scholar] [PubMed]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The old world and new world alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef]
- Kim, Y.D.; Reynaud, J.M.; Rasalouskaya, A.; Akhrymuk, I.; Mobley, J.A.; Frolov, I.; Frolova, E.I. New world and old world alphaviruses have evolved to exploit different components of stress granules, FXR and G3BP proteins, for assembly of viral replication complexes. PLoS Pathog. 2016, 12, e1005810. [Google Scholar] [CrossRef]
- Fros, J.J.; Geertsema, C.; Zouache, K.; Baggen, J.; Domeradzka, N.; van Leeuwen, D.M.; Flipse, J.; Vlak, J.M.; Failloux, A.B.; Pijlman, G.P. Mosquito Rasputin interacts with chikungunya virus nsP3 and determines the infection rate in Aedes albopictus. Parasit. Vectors 2015, 8, 464. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus infection: Host cell shut-off and inhibition of antiviral responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- Mutso, M.; Morro, A.; Smedberg, C.; Kasvandik, S.; Aquilimeba, M.; Teppor, M.; Tarve, L.; Lulla, A.; Lulla, V.; Saul, S. Mutation of CD2AP and SH3KBP1 binding motif in alphavirus nsP3 hypervariable domain results in attenuated virus. Viruses 2018, 10, 226. [Google Scholar] [CrossRef]
- Varjak, M.; Zusinaite, E.; Merits, A. Novel functions of the alphavirus nonstructural protein nsP3 C-terminal region. J. Virol. 2010, 84, 2352–2364. [Google Scholar] [CrossRef] [PubMed]
- Meshram, D.C.; Shiliaev, N.; Frolova, E.I.; Frolov, I. Hypervariable domain of nsP3 of Eastern Equine Encephalitis virus is a critical determinant of viral virulence. J. Virol. 2020, 94, e00617–e00620. [Google Scholar] [CrossRef]
- Lark, T.; Keck, F.; Narayanan, A. Interactions of Alphavirus nsP3 protein with host proteins. Front. Microbiol. 2017, 8, 2652. [Google Scholar] [CrossRef]
- Aloha, T.; Merits, A. Functions of Chikungunya virus nonstructural proteins. In Chikungunya Virus: Advances in Biology, Pathogenesis, and Treatment; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Malet, H.; Coutard, B.; Jamal, S.; Dutartre, H.; Papageorgiou, N.; Neuvonen, M.; Ahola, T.; Forrester, N.; Gould, E.A.; Lafitte, D.; et al. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 2009, 83, 6534–6545. [Google Scholar] [CrossRef]
- Creighton, T.E. Proteins: Structures and Molecular Properties, 2nd ed.; W.H. Freeman and Company: New York, NY, USA, 1993. [Google Scholar]
- Agback, P.; Shernyukov, A.; Dominguez, F.; Agback, T.; Frolova, E.I. Novel NMR assignment strategy reveals structural heterogeneity in solution of the nsP3 HVD domain of Venezuelan Equine Encephalitis virus. Molecules 2020, 25, 5824. [Google Scholar] [CrossRef]
- Agback, P.; Dominguez, F.; Pustovalova, Y.; Lukash, T.; Shiliaev, N.; Orekhov, V.Y.; Frolov, I.; Agback, T.; Frolova, E.I. Structural characterization and biological function of bivalent binding of CD2AP to intrinsically disordered domain of Chikungunya virus nsP3 protein. Virology 2019, 537, 130–142. [Google Scholar] [CrossRef]
- Bakar, A.F.; Ng, L.F.P. Nonstructural proteins of alphavirus-potential targets for drug development. Viruses 2018, 10, 71. [Google Scholar] [CrossRef]
- Tuittila, M.; Hinkkanen, A.E. Amino acid mutations in the replicase protein nsP3 of Semliki Forest virus cumulatively affect neurovirulence. J. Gen. Virol. 2003, 84, 1525–1533. [Google Scholar] [CrossRef]
- Burt, J.F.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Weaver, C.S.; Winegar, R.; Manger, I.D.; Forrester, N.L. Alphaviruses: Population genetics and determinants of emergence. Antivir. Res. 2012, 94, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Frolov, I. Topley and Wilson’s Microbiology and Microbial Infections: Virology; ASM Press: Washington, DC, USA, 2005. [Google Scholar]
- Chapter 28—Togaviridae. In Fenner’s Veterinary Virology, 5th ed.; MacLachlan, N.J., Dubovi, E.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 511–524. [Google Scholar]
- Ng, L.F.P. Immunopathology of Chikungunya virus infection: Lessons learned from patients and animal models. Annu. Rev. Virol. 2017, 4, 413–427. [Google Scholar] [CrossRef]
- Ryman, D.K.; Klimstra, W.B. Host responses to alphavirus infection. Immunol. Rev. 2008, 225, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Fields, N.B.; Knipe, D.M.; Griffin, D.E. Fields Virology; Lippincott-Raven: Philadelphia, PA, USA, 2001. [Google Scholar]
- Karpf, R.A.; Blake, J.M.; Brown, D.T. Characterization of the infection of Aedes albopictus cell clones by Sindbis virus. Virus Res. 1997, 50, 1–13. [Google Scholar] [CrossRef]
- Mudiganti, U.; Hernandez, R.; Ferreira, D.; Brown, D.T. Sindbis virus infection of two model insect cell systems—A comparative study. Virus Res. 2006, 122, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; Sanz, M.A.; González-Almela, E. The regulation of translation in alphavirus-infected cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, R. Chikungunya virus and (Re-)emerging alphaviruses. Viruses 2019, 11, 779. [Google Scholar] [CrossRef]
- Roberts, C.G.; Zothner, C.; Remenyi, R.; Merits, A.; Stonehouse, N.J.; Harris, M. Evaluation of a range of mammalian and mosquito cell lines for use in Chikungunya virus research. Sci. Rep. 2017, 7, 14641. [Google Scholar] [CrossRef]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef]
- Strauss, G.E.; Rice, C.M.; Strauss, J.H. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 1984, 133, 92–110. [Google Scholar] [CrossRef]
- Aaskov, J.; Jones, A.; Choi, W.; Lowry, K.; Stewart, E. Lineage replacement accompanying duplication and rapid fixation of an RNA element in the nsP3 gene in a species of alphavirus. Virology 2011, 410, 353–359. [Google Scholar] [CrossRef]
- Metz, W.S.; Pijlman, G.P. Function of Chikungunya virus structural proteins. In Chikungunya Virus: Advances in Biology, Pathogenesis, and Treatment; Okeoma, C.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 63–74. [Google Scholar]
- Firth, E.A.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef]
- Jenkins, M.G.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef]
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of Virology. Volume I: Molecular Biology, 3rd ed.; ASM Press: Washington, DC, USA, 2009. [Google Scholar]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.S.; Wong, S.-C.; Kwek, D.J.C.; Tolou, H.; Lin, R.T.P.; Tambyah, P.A.; Rénia, L.; et al. Active infection of human blood monocytes by chikungunya virus triggers an innate immune response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; le Roux, K.; Prevost, M.-C.; Fsihi, H.; et al. Characterization of reemerging chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef]
- Das, T.; Jaffar-Bandjee, M.C.; Hoarau, J.J.; Trotot, P.K.; Denizot, M.; Lee-Pat-Yuen, G.; Sahoo, R.; Guiraud, P.; Ramful, D.; Robin, S. Chikungunya fever: CNS infection and pathologies of a re-emerging arbovirus. Prog. Neurobiol. 2010, 91, 121–129. [Google Scholar] [CrossRef]
- Van Duijl-Richter, M.K.; Hoornweg, T.E.; Rodenhuis-Zybert, I.A.; Smit, J.M. Early events in chikungunya virus infection—From virus cell binding to membrane fusion. Viruses 2015, 7, 3647. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Adsorptive endocytosis of Semliki Forest virus. J. Mol. Biol. 1980, 142, 439–454. [Google Scholar] [CrossRef]
- Smith, A.L.; Tignor, G.H. Host cell receptors for two strains of sindbis virus. Arch. Virol. 1980, 66, 11–26. [Google Scholar] [CrossRef]
- Klimstra, W.B.; Nangle, E.M.; Smith, M.S.; Yurochko, A.D.; Ryman, K.D. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 2003, 77, 12022–12032. [Google Scholar] [CrossRef]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [CrossRef]
- Malygin, A.; Bondarenko, E.I.; Ivanisenko, V.A.; Protopopova, E.V.; Karpova, G.G.; Loktev, V.B. C-terminal fragment of human laminin-binding protein contains a receptor domain for Venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry 2009, 74, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural resistance-associated macrophage protein is a cellular receptor for Sindbis virus in both insect and mammalian hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, L.; Yang, Y.; Jia, J.; Fu, S.; Feng, Y.; He, Y.; Li, J.-P.; Liang, G. Interaction of E2 glycoprotein with heparan sulfate is crucial for cellular infection of Sindbis virus. PLoS ONE 2010, 5, e9656. [Google Scholar] [CrossRef]
- Gardner, C.L.; Ebel, G.D.; Ryman, K.D.; Klimstra, W.B. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc. Natl. Acad. Sci. USA 2011, 108, 16026–16031. [Google Scholar] [CrossRef]
- Klimstra, W.B.; Ryman, K.D.; Johnston, R.E. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 1998, 72, 7357–7366. [Google Scholar] [CrossRef]
- Byrnes, A.P.; Griffin, D.E. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 1998, 72, 7349–7356. [Google Scholar] [CrossRef]
- Bernard, K.A.; Klimstra, W.B.; Johnston, R.E. Mutations in the E2 glycoprotein of venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 2000, 276, 93–103. [Google Scholar] [CrossRef]
- Heil, M.L.; Albee, A.; Strauss, J.H.; Kuhn, R.J. An amino acid substitution in the coding region of the e2 glycoprotein adapts ross river virus to utilize heparan sulfate as an attachment moiety. J. Virol. 2001, 75, 6303–6309. [Google Scholar] [CrossRef]
- La Linn, M.; Eble, J.A.; Lubken, C.; Slade, R.W.; Heino, J.; Davies, J.; Suhrbier, A. An arthritogenic alphavirus uses the al-pha1beta1 integrin collagen receptor. Virology 2005, 336, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.-Y.; Panyasrivanit, M.; et al. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-San Martin, C.; Sosa, H.; Kielian, M. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 2008, 4, 600–608. [Google Scholar] [CrossRef]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The fusion glycoprotein shell of Semliki forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef]
- DeTulleo, L.; Kirchhausen, T. The clathrin endocytic pathway in viral infection. EMBO J. 1998, 17, 4585–4593. [Google Scholar] [CrossRef]
- Lee, R.C.H.; Hapuarachchi, H.C.; Chen, K.C.; Hussain, K.M.; Chen, H.; Low, S.L.; Ng, L.C.; Lin, R.; Ng, M.M.-L.; Chu, J.J.H. Mosquito cellular factors and functions in mediating the infectious entry of Chikungunya virus. PLoS Negl. Trop. Dis. 2013, 7, e2050. [Google Scholar] [CrossRef]
- Bernard, E.; Solignat, M.; Gay, B.; Chazal, N.; Higgs, S.; Devaux, C.; Briant, L. Endocytosis of Chikungunya virus into mammalian cells: Role of clathrin and early endosomal compartments. PLoS ONE 2010, 5, e11479. [Google Scholar] [CrossRef]
- Vasiljeva, L.; Valmu, L.; Kääriäinen, L.; Merits, A. Site-specific protease activity of the carboxyl-terminal domain of Semliki forest virus replicase protein nsP2. J. Biol. Chem. 2001, 276, 30786–30793. [Google Scholar] [CrossRef]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef]
- Laakkonen, P.; Ahola, T.; Kääriäinen, L. The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J. Biol. Chem. 1996, 271, 28567–28571. [Google Scholar] [CrossRef]
- Laakkonen, P.; Auvinen, P.; Kujala, P.; Kaariainen, L. Alphavirus replicase protein NSP1 induces filopodia and rear-rangement of actin filaments. J. Virol. 1998, 72, 10265–10269. [Google Scholar] [CrossRef]
- Fros, J.J.; Van Der Maten, E.; Vlak, J.M.; Pijlman, G.P. The C-terminal domain of Chikungunya virus nsP2 independently governs viral RNA replication, cytopathicity, and inhibition of interferon signaling. J. Virol. 2013, 87, 10394–10400. [Google Scholar] [CrossRef]
- Lemm, A.J.; Rice, C.M. Roles of nonstructural polyproteins and cleavage products in regulating Sindbis virus RNA repli-cation and transcription. J. Virol. 1993, 67, 1916–1926. [Google Scholar] [CrossRef]
- Mai, J.; Sawicki, S.G.; Sawicki, D.L. Fate of minus-strand templates and replication complexes produced by a P23-cleavage-defective mutant of Sindbis virus. J. Virol. 2009, 83, 8553–8564. [Google Scholar] [CrossRef]
- Utt, A.; Das, P.K.; Varjak, M.; Lulla, V.; Lulla, A.; Merits, A. Mutations conferring a noncytotoxic phenotype on Chikungunya virus replicons compromise enzymatic properties of nonstructural protein 2. J. Virol. 2014, 89, 3145–3162. [Google Scholar] [CrossRef]
- Spuul, P.; Balistreri, G.; Kääriäinen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki forest virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef]
- Wu, J.Q.H. Virulence determinants of New World alphaviruses and broad-acting therapeutic strategies. Future Virol. 2015, 10, 647–657. [Google Scholar] [CrossRef]
- Button, J.M.; Qazi, S.A.; Wang, J.C.-Y.; Mukhopadhyay, S. Revisiting an old friend: New findings in alphavirus structure and assembly. Curr. Opin. Virol. 2020, 45, 25–33. [Google Scholar] [CrossRef]
- Ramsey, J.; Mukhopadhyay, S. Disentangling the frames, the state of research on the alphavirus 6K and TF proteins. Viruses 2017, 9, 228. [Google Scholar] [CrossRef]
- Neuvonen, M.; Kazlauskas, A.; Martikainen, M.; Hinkkanen, A.; Ahola, T.; Saksela, K. SH3 domain-mediated recruitment of host cell amphiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog. 2011, 7, e1002383. [Google Scholar] [CrossRef] [PubMed]
- Hahn, Y.S.; Strauss, E.G.; Strauss, J.H. Mapping of RNA-temperature-sensitive mutants of Sindbis virus: Assignment of complementation groups A, B, and G to nonstructural proteins. J. Virol. 1989, 63, 3142–3150. [Google Scholar] [CrossRef] [PubMed]
- LaStarza, M.W.; Lemm, J.A.; Rice, C.M. Genetic analysis of the nsP3 region of Sindbis virus: Evidence for roles in mi-nus-strand and subgenomic RNA synthesis. J. Virol. 1994, 68, 5781–5791. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Sawicki, S.G.; Sawicki, D.L. Alphavirus nsP3 functions to form replication complexes transcribing negative-strand RNA. J. Virol. 1994, 68, 6466–6475. [Google Scholar] [CrossRef] [PubMed]
- Cristea, I.M.; Carroll, J.W.; Rout, M.P.; Rice, C.M.; Chait, B.T.; MacDonald, M.R. Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 2006, 281, 30269–30278. [Google Scholar] [CrossRef] [PubMed]
- Després, P.; Griffin, J.W.; Griffin, D.E. Effects of anti-E2 monoclonal antibody on sindbis virus replication in AT3 cells expressing bcl-2. J. Virol. 1995, 69, 7006–7014. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.; Gorchakov, R.; Garmashova, N.; Atasheva, S.; Vergara, L.A.; Frolov, I. Formation of nsP3-specific protein complexes during Sindbis virus replication. J. Virol. 2006, 80, 4122–4134. [Google Scholar] [CrossRef]
- Gorchakov, R.; Garmashova, N.; Frolova, E.; Frolov, I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 2008, 82, 10088–10101. [Google Scholar] [CrossRef]
- Frolov, I.; Kim, D.Y.; Akhrymuk, M.; Mobley, J.A.; Frolova, E.I. Hypervariable domain of Eastern Equine Encephalitis virus nsP3 redundantly utilizes multiple cellular proteins for replication complex assembly. J. Virol. 2017, 91, e00371-17. [Google Scholar] [CrossRef]
- De, I.; Sawicki, S.G.; Sawicki, D.L. Sindbis virus RNA-negative mutants that fail to convert from minus-strand to plus-strand synthesis: Role of the nsP2 protein. J. Virol. 1996, 70, 2706–2719. [Google Scholar] [CrossRef]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kääriäinen, L. Elimination of phosphorylation sites of Semliki forest virus replicase protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef]
- Johnston, R.; Peters, C. Fields Virology, 3rd ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; Volume I, pp. 843–898. [Google Scholar]
- Van Regenmortel, M.H.; Fauquet, C.M.; Bishop, D.H.; Carstens, E.; Estes, M.; Lemon, S.; Maniloff, J.; Mayo, M.; McGeoch, D.; Pringle, C. Virus Taxonomy: Classification and Nomenclature of Viruses. Seventh Report of the International Committee on Taxonomy of Viruses; Academic Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Ferro, C.; Boshell, J.; Moncayo, A.C.; Gonzalez, M.; Ahumada, M.L.; Kang, W.; Weaver, S.C. Natural enzootic vectors of Venezuelan equine encephalitis virus, Magdalena Valley, Colombia. Emerg. Infect. Dis. 2003, 9, 49–54. [Google Scholar] [CrossRef]
- Reeves, L.E.; Hoyer, I.; Acevedo, C.; Burkett-Cadena, N.D. Host associations of Culex (Melanoconion) atratus (Diptera: Culicidae) and Culex (Melanoconion) pilosus from Florida, USA. Insects 2019, 10, 239. [Google Scholar] [CrossRef]
- Saxton-Shaw, K.D.; Ledermann, J.P.; Borland, E.; Stovall, J.L.; Mossel, E.C.; Singh, A.J.; Wilusz, J.; Powers, A.M. O’nyong nyong virus molecular determinants of unique vector specificity reside in non-structural protein 3. PLoS Negl. Trop. Dis. 2013, 7, e1931. [Google Scholar] [CrossRef]
- Atkins, G.J. The pathogenesis of Alphaviruses. ISRN Virol. 2013, 2013, 861912. [Google Scholar] [CrossRef]
- Göertz, G.P.; Lingemann, M.; Geertsema, C.; Abma-Henkens, M.H.C.; Vogels, C.B.F.; Koenraadt, C.J.M.; Van Oers, M.M.; Pijlman, G.P. Conserved motifs in the hypervariable domain of chikungunya virus nsP3 required for transmission by Aedes aegypti mosquitoes. PLOS Negl. Trop. Dis. 2018, 12, e0006958. [Google Scholar] [CrossRef]
- Flint, J.; Racaniello, V.R.; Rall, G.F.; Skalka, A.M. Principles of Virology, 4th ed.; American Society of Microbiology: Washington, DC, USA, 2015. [Google Scholar]
- Weger-Lucarelli, J.; Aliota, M.T.; Wlodarchak, N.; Kamlangdee, A.; Swanson, R.; Osorio, J.E. Dissecting the role of E2 protein domains in alphavirus pathogenicity. J. Virol. 2015, 90, 2418–2433. [Google Scholar] [CrossRef]
- Tucker, P.C.; Griffin, D.E. Mechanism of altered Sindbis virus neurovirulence associated with a single-amino-acid change in the E2 Glycoprotein. J. Virol. 1991, 65, 1551–1557. [Google Scholar] [CrossRef]
- Tucker, P.C.; Lee, S.H.; Bui, N.; Martinie, D.; Griffin, D.E. Amino acid changes in the Sindbis virus E2 glycoprotein that increase neurovirulence improve entry into neuroblastoma cells. J. Virol. 1997, 71, 6106–6112. [Google Scholar] [CrossRef]
- Atkins, G.J.; Sheahan, B.J. Molecular determinants of alphavirus neuropathogenesis in mice. J. Gen. Virol. 2016, 97, 1283–1296. [Google Scholar] [CrossRef]
- Kinney, R.M.; Chang, G.J.; Tsuchiya, K.R.; Sneider, J.M.; Roehrig, J.T.; Woodward, T.M.; Trent, D.W. Attenuation of Venezuelan equine encephalitis virus strain TC-83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J. Virol. 1993, 67, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Powell, N.; Greenwald, G.F.; Willis, L.V.; Johnson, B.J.; Smith, J.F.; Johnston, R.E. Attenuating mutations in the E2 glycoprotein gene of Venezuelan equine encephalitis virus: Construction of single and multiple mutants in a full-length cDNA clone. Virology 1991, 183, 20–31. [Google Scholar] [CrossRef]
- Aguilar, P.V.; Leung, L.W.; Wang, E.; Weaver, S.C.; Basler, C.F. A five-amino-acid deletion of the Eastern Equine Encephalitis virus capsid protein attenuates replication in mammalian systems but not in mosquito cells. J. Virol. 2008, 82, 6972–6983. [Google Scholar] [CrossRef] [PubMed]
- Peltier, D.C.; LaZear, H.M.; Farmer, J.R.; Diamond, M.S.; Miller, D.J. Neurotropic arboviruses induce interferon regulatory factor 3-mediated neuronal responses that are cytoprotective, interferon independent, and inhibited by Western Equine Encephalitis virus capsid. J. Virol. 2012, 87, 1821–1833. [Google Scholar] [CrossRef]
- Reynaud, J.M.; Atasheva, S.; Rasalouskaya, A.; White, J.P.; Diamond, M.S.; Weaver, S.C.; Frolova, E.I.; Frolov, I. IFIT1 dif-ferentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLoS Pathog. 2015, 11, e1004863. [Google Scholar] [CrossRef]
- Fros, J.J.; Liu, W.J.; Prow, N.A.; Geertsema, C.; Ligtenberg, M.; VanLandingham, D.L.; Schnettler, E.; Vlak, J.M.; Suhrbier, A.; Khromykh, A.A.; et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 2010, 84, 10877–10887. [Google Scholar] [CrossRef]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; Van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef]
- Fazakerley, J.K.; Boyd, A.; Mikkola, M.L.; Kääriäinen, L. A single amino acid change in the nuclear localization sequence of the nsP2 protein affects the neurovirulence of Semliki forest virus. J. Virol. 2002, 76, 392–396. [Google Scholar] [CrossRef]
- Heise, M.T.; Simpson, D.A.; Johnston, R.E. A single amino acid change in nsP1 attenuates neurovirulence of the Sind-bis-group alphavirus S.A.AR86. J. Virol. 2000, 74, 4207–4213. [Google Scholar] [CrossRef]
- Suthar, M.S.; Shabman, R.; Madric, K.; Lambeth, C.; Heise, M.T. Identification of adult mouse neurovirulence determinants of the Sindbis virus strain AR86. J. Virol. 2005, 79, 4219–4228. [Google Scholar] [CrossRef]
- Logue, C.H.; Sheahan, B.J.; Atkins, G.J. The 5′ untranslated region as a pathogenicity determinant of Semliki Forest virus in mice. Virus Genes 2008, 36, 313–321. [Google Scholar] [CrossRef]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A Viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef]
- Kulasegaran-Shylini, R.; Thiviyanathan, V.; Gorenstein, D.G.; Frolov, I. The 5′UTR-specific mutation in VEEV TC-83 genome has a strong effect on RNA replication and subgenomic RNA synthesis, but not on translation of the encoded proteins. Virology 2009, 387, 211–221. [Google Scholar] [CrossRef]
- Tuittila, M.T.; Santagati, M.G.; Röyttä, M.; Määttä, J.A.; Hinkkanen, A.E. Replicase complex genes of Semliki Forest virus confer lethal neurovirulence. J. Virol. 2000, 74, 4579–4589. [Google Scholar] [CrossRef]
- Saul, S.; Ferguson, M.C.; Cordonin, C.; Fragkoudis, R.; Ool, M.; Tamberg, N.; Sherwood, K.; Fazakerley, J.; Merits, A. Differences in processing determinants of nonstructural polyprotein and in the sequence of nonstructural protein 3 affect neurovirulence of Semliki forest virus. J. Virol. 2015, 89, 11030–11045. [Google Scholar] [CrossRef]
- Meshram, C.; Phillips, A.T.; Lukash, T.; Shiliaev, N.; Frolova, E.I.; Frolov, I. Mutations in hypervariable domain of Venezuelan Equine Encephalitis virus nsP3 protein differentially affect viral replication. J. Virol. 2019, 94, e01841-19. [Google Scholar] [CrossRef]
- McPherson, R.L.; Abraham, R.; Sreekumar, E.; Ong, S.-E.; Cheng, S.-J.; Baxter, V.K.; Kistemaker, H.A.V.; Filippov, D.V.; Griffin, D.E.; Leung, A.K.L. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc. Natl. Acad. Sci. USA 2017, 114, 1666–1671. [Google Scholar] [CrossRef]
- Abraham, R.; Hauer, D.; McPherson, R.L.; Utt, A.; Kirby, I.T.; Cohen, M.S.; Merits, A.; Leung, A.K.L.; Griffin, D.E. ADP-ribosyl–binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, e10457–e10466. [Google Scholar] [CrossRef]
- Park, E.; Griffin, D.E. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef]
- Dé, I.; Fata-Hartley, C.; Sawicki, S.G.; Sawicki, D.L. Functional analysis of nsP3 phosphoprotein mutants of SindbisVirus. J. Virol. 2003, 77, 13106–13116. [Google Scholar] [CrossRef]
- Abraham, R.; McPherson, R.L.; Dasovich, M.; Badiee, M.; Leung, A.K.L.; Griffin, D.E. Both ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain affect neurovirulence in mice. mBio 2020, 11, e03253-19. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, S.; Sheahan, B.J.; Atkins, G.J. Deletions in the hypervariable domain of the nsP3 gene attenuate Semliki Forest virus virulence. J. Gen. Virol. 2006, 87, 937–947. [Google Scholar] [CrossRef]
- Myles, K.M.; Kelly, C.L.H.; Ledermann, J.P.; Powers, A.M. Effects of an opal termination codon preceding the nsP4 gene sequence in the O’Nyong-Nyong virus genome on Anopheles gambiae infectivity. J. Virol. 2006, 80, 4992–4997. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Ludwig, M.L.; Rwaguma, E.B.; Lutwama, J.J.; Kram, T.M.; Karabatsos, N.; Cropp, B.C.; Miller, B.R. Emergence of epidemic O’nyong-nyong Fever in Uganda after a 35-year absence: Genetic characterization of the virus. Virology 1998, 252, 258–268. [Google Scholar] [CrossRef]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef]
- Jones, J.; Long, K.M.; Whitmore, A.C.; Sanders, W.; Thurlow, L.R.; Brown, J.; Morrison, C.R.; Vincent, H.; Peck, K.M.; Browning, C.; et al. Disruption of the opal stop codon attenuates Chikungunya virus-induced arthritis and pathology. mBio 2017, 8, e01456-17. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Ahola, T.; McInerney, G.M. The C-terminal repeat domains of nsP3 from the Old World alphaviruses bind directly to G3BP. J. Virol. 2014, 88, 5888–5893. [Google Scholar] [CrossRef]
- Park, E.; Griffin, D.E. Interaction of Sindbis virus non-structural protein 3 with poly (ADP-ribose) polymerase 1 in neuronal cells. J. Gen. Virol. 2009, 90, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Nowee, G.; Bakker, J.W.; Geertsema, C.; Ros, V.I.D.; Göertz, G.P.; Fros, J.J.; Pijlman, G.P. A tale of 20 alphaviruses; inter-species diversity and conserved interactions between viral non-structural protein 3 and stress granule proteins. Front. Cell Dev. Biol. 2021, 9, 625711. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Hedestam, G.B.K.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into Cytoplasmic Foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Jones, A.; Lowry, K.; Aaskov, J.; Holmes, E.C.; Kitchen, A. Molecular evolutionary dynamics of Ross River virus and implications for vaccine efficacy. J. Gen. Virol. 2009, 91, 182–188. [Google Scholar] [CrossRef]
- Michie, A.; Dhanasekaran, V.; Lindsay, M.D.A.; Neville, P.J.; Nicholson, J.; Jardine, A.; MacKenzie, J.S.; Smith, D.W.; Imrie, A. Genome-scale phylogeny and evolutionary analysis of Ross River virus reveals periodic sweeps of lineage dominance in Western Australia, 1977–2014. J. Virol. 2019, 94, e01234-19. [Google Scholar] [CrossRef]
- Foo, S.S.; Chen, W.; Herrero, L.; Bettadapura, J.; Narayan, J.; Dar, L.; Broor, S.; Mahalingam, S. The genetics of alphaviruses. Future Virol. 2011, 6, 1407–1422. [Google Scholar] [CrossRef]
- Althouse, B.M.; Hanley, K.A. The tortoise or the hare? Impacts of within-host dynamics on transmission success of arthropod-borne viruses. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140299. [Google Scholar] [CrossRef]
- Salonen, A.; Vasiljeva, L.; Merits, A.; Magden, J.; Jokitalo, E.; Kääriäinen, L. Properly folded nonstructural polyprotein directs the Semliki forest virus replication complex to the endosomal compartment. J. Virol. 2003, 77, 1691–1702. [Google Scholar] [CrossRef]
- Lampio, A.; Kilpeläinen, I.; Pesonen, S.; Karhi, K.; Auvinen, P.; Somerharju, P.; Kääriäinen, L. Membrane binding mechanism of an RNA virus-capping enzyme. J. Biol. Chem. 2000, 275, 37853–37859. [Google Scholar] [CrossRef]
- Spuul, P.; Salonen, A.; Merits, A.; Jokitalo, E.; Kääriäinen, L.; Ahola, T. Role of the amphipathic peptide of semliki forest virus replicase Protein nsP1 in membrane association and virus replication. J. Virol. 2006, 81, 872–883. [Google Scholar] [CrossRef]
- Ahola, T.; Kujala, P.; Tuittila, M.; Blom, T.; Laakkonen, P.; Hinkkanen, A.; Auvinen, P. Effects of palmitoylation of replicase protein nsP1 on alphavirus infection. J. Virol. 2000, 74, 6725–6733. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.F.; Sawicki, S.G.; Sawicki, D.L. Sindbis virus nsP1 functions in negative-strand RNA synthesis. J. Virol. 1991, 65, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Durbin, R.; Huang, H.V.; Rice, C.M.; Stollar, V. Association of the Sindbis virus RNA methyltransferase activity with the nonstructural protein nsP1. Virology 1989, 170, 385–391. [Google Scholar] [CrossRef]
- Li, C.; Guillén, J.; Rabah, N.; Blanjoie, A.; Debart, F.; Vasseur, J.-J.; Canard, B.; Decroly, E.; Coutard, B. mRNA capping by Venezuelan Equine Encephalitis virus nsP1: Functional characterization and implications for antiviral research. J. Virol. 2015, 89, 8292–8303. [Google Scholar] [CrossRef]
- Ahola, T.; Kaariainen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef]
- Ramakrishnan, C.; Kutumbarao, N.H.V.; Suhitha, S.; Velmurugan, D. Structure-function relationship of Chikungunya nsP2 protease: A comparative study with papain. Chem. Biol. Drug Des. 2017, 89, 772–782. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Aher, P.P.; Lole, K.S. NTPase and 5′-RNA triphosphatase activities of Chikungunya virus nsP2 protein. PLoS ONE 2011, 6, e22336. [Google Scholar] [CrossRef]
- Das, P.K.; Merits, A.; Lulla, A. Functional cross-talk between distant domains of Chikungunya virus non-structural protein 2 is decisive for its RNA-modulating activity. J. Biol. Chem. 2014, 289, 5635–5653. [Google Scholar] [CrossRef]
- Rausalu, K.; Utt, A.; Quirin, T.; Varghese, F.S.; Žusinaite, E.; Das, P.K.; Ahola, T.; Merits, A. Chikungunya virus infectivity, RNA replication and non-structural polyprotein processing depend on the nsP2 protease’s active site cysteine residue. Sci. Rep. 2016, 6, 37124. [Google Scholar] [CrossRef]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [CrossRef]
- Rubach, J.K.; Wasik, B.R.; Rupp, J.C.; Kuhn, R.J.; Hardy, R.W.; Smith, J.L. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology 2009, 384, 201–208. [Google Scholar] [CrossRef]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef]
- Lykouras, M.V.; Tsika, A.C.; Lichiere, J.; Papageorgiou, N.; Coutard, B.; Bentrop, D.; Spyroulias, G.A. NMR study of non-structural proteins-part III: (1)H, (13)C, (15)N backbone and side-chain resonance assignment of macro domain from Chikungunya virus (CHIKV). Biomol. NMR Assign. 2018, 12, 31–35. [Google Scholar] [CrossRef]
- Makrynitsa, G.I.; Ntonti, D.; Marousis, K.D.; Tsika, A.C.; Lichiere, J.; Papageorgiou, N.; Coutard, B.; Bentrop, D.; Spyroulias, G.A. NMR study of non-structural proteins—Part II: (1)H, (13)C, (15)N backbone and side-chain resonance assignment of macro domain from Venezuelan equine encephalitis virus (VEEV). Biomol. NMR Assign. 2015, 9, 247–251. [Google Scholar] [CrossRef]
- Melekis, E.; ATsika, C.; Lichiere, J.; Chasapis, C.T.; Margiolaki, I.; Papageorgiou, N.; Coutard, B.; Bentrop, D.; Spyroulias, G.A. NMR study of non-structural proteins—Part I: (1)H, (13)C, (15)N backbone and side-chain resonance assignment of macro domain from Mayaro virus (MAYV). Biomol. NMR Assign. 2015, 9, 191–195. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef]
- Beitzel, B.F.; Bakken, R.R.; Smith, J.M.; Schmaljohn, C.S. High-resolution functional mapping of the Venezuelan Equine Encephalitis virus genome by insertional mutagenesis and massively parallel sequencing. PLoS Pathog. 2010, 6, e1001146. [Google Scholar] [CrossRef]
- Rungrotmongkol, T.; Nunthaboot, N.; Malaisree, M.; Kaiyawet, N.; Yotmanee, P.; Meeprasert, A.; Hannongbua, S. Molecular insight into the specific binding of ADP-ribose to the nsP3 macro domains of chikungunya and Venezuelan equine encephalitis viruses: Molecular dynamics simulations and free energy calculations. J. Mol. Graph. Model 2010, 29, 347–353. [Google Scholar] [CrossRef]
- Yusof, M.; Wasi Ahmad, N.; Azleen, Z.; Vythilingam, I.; Yusuf, N.A.; Azahari, A.; Saat, Z.; Hanlim, L. The first isolation of Chikungunya virus from non-human primates in Malaysia. J. Gen. Mol. Virol. 2009, 1, 35–39. [Google Scholar]
- Foy, N.J.; Akhrymuk, M.; Akhrymuk, I.; Atasheva, S.; Bopda-Waffo, A.; Frolov, I.; Frolova, E.I. Hypervariable domains of nsP3 proteins of new world and old world alphaviruses mediate formation of distinct, virus-specific protein complexes. J. Virol. 2012, 87, 1997–2010. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Kustatscher, G.; Buhecha, H.R.; Allen, M.D.; Pugieux, C.; Sait, F.; Bycroft, M.; Ladurner, A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Feijs, K.L.; Forst, A.H.; Verheugd, P.; Luscher, B. Macrodomain-containing proteins: Regulating new intra-cellular functions of mono (ADP-ribosyl) ation. Nat. Rev. Mol. Cell Biol. 2013, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral macro domains reverse protein ADP-Ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef]
- Puranik, N.V.; Rani, R.; Singh, V.A.; Tomar, S.; Puntambekar, H.M.; Srivastava, P. Evaluation of the antiviral potential of halogenated dihydrorugosaflavonoids and molecular modeling with nsP3 protein of Chikungunya virus (CHIKV). ACS Omega 2019, 4, 20335–20345. [Google Scholar] [CrossRef]
- Lulla, A.; Lulla, V.; Merits, A. Macromolecular assembly-driven processing of the 2/3 cleavage site in the alphavirus replicase polyprotein. J. Virol. 2012, 86, 553–565. [Google Scholar] [CrossRef]
- Egloff, M.-P.; Malet, H.; Putics, Á.; Heinonen, M.; Dutartre, H.; Frangeul, A.; Gruez, A.; Campanacci, V.; Cambillau, C.; Ziebuhr, J. Structural and functional basis for ADP-ribose and poly (ADP-ribose) binding by viral macro domains. J. Virol. 2006, 80, 8493–8502. [Google Scholar] [CrossRef]
- Fehr, A.R.; Jankevicius, G.; Ahel, I.; Perlman, S. Viral macrodomains: Unique mediators of viral replication and pathogenesis. Trends Microbiol. 2018, 26, 598–610. [Google Scholar] [CrossRef]
- Michel, G.; Petrakova, O.; Atasheva, S.; Frolov, I. Adaptation of Venezuelan equine encephalitis virus lacking 51-nt conserved sequence element to replication in mammalian and mosquito cells. Virology 2007, 362, 475–487. [Google Scholar] [CrossRef]
- Foy, N.J.; Akhrymuk, M.; Shustov, A.V.; Frolova, E.I.; Frolov, I. Hypervariable domain of nonstructural protein nsP3 of Venezuelan Equine Encephalitis virus determines cell-specific mode of virus replication. J. Virol. 2013, 87, 7569–7584. [Google Scholar] [CrossRef]
- Gao, Y.; Goonawardane, N.; Ward, J.; Tuplin, A.; Harris, M. Multiple roles of the non-structural protein 3 (nsP3) alphavirus unique domain (AUD) during Chikungunya virus genome replication and transcription. PLoS Pathog. 2019, 15, e1007239. [Google Scholar] [CrossRef]
- Makrynitsa, G.I.; Ntonti, D.; Marousis, K.D.; Birkou, M.; Matsoukas, M.-T.; Asami, S.; Bentrop, D.; Papageorgiou, N.; Canard, B.; Coutard, B.; et al. Conformational plasticity of the VEEV macro domain is important for binding of ADP-ribose. J. Struct. Biol. 2019, 206, 119–127. [Google Scholar] [CrossRef]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.; Ahel, I.; Perlman, S. The conserved Coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome Coronavirus infection. mBio 2016, 7, e01721-16. [Google Scholar] [CrossRef]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Götte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G. Differential phosphatidylinositol-3-kinase-Akt-mTOR activation by Semliki Forest and chikungunya viruses is dependent on nsP3 and connected to replication complex internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef]
- Kumar, R.; Mehta, D.; Mishra, N.; Nayak, D.; Sunil, S. Role of host-mediated post-translational modifications (PTMs) in RNA virus pathogenesis. Int. J. Mol. Sci. 2021, 22, 323. [Google Scholar] [CrossRef]
- Teppor, M.; Žusinaite, E.; Merits, A. Phosphorylation sites in the hypervariable domain in chikungunya virus nsP3 are crucial for viral replication. J. Virol. 2021, e02276-20. [Google Scholar] [CrossRef]
- Frolov, I.; Frolova, E.I. Molecular virology of Chikungunya virus. Curr. Top. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Hellström, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef]
- Wong, K.Z.; Chu, J.J.H. The interplay of viral and host factors in chikungunya virus infection: Targets for antiviral strategies. Viruses 2018, 10, 294. [Google Scholar] [CrossRef]
- Schulte, T.; Liu, L.; Panas, M.D.; Thaa, B.; Dickson, N.; Götte, B.; Achour, A.; McInerney, G.M. Combined structural, biochemical and cellular evidence demonstrates that both FGDF motifs in alphavirus nsP3 are required for efficient replication. Open Biol. 2016, 6, 160078. [Google Scholar] [CrossRef]
- Meertens, L.; Hafirassou, M.L.; Couderc, T.; Bonnet-Madin, L.; Kril, V.; Kümmerer, B.M.; Labeau, A.; Brugier, A.; Simon-Loriere, E.; Burlaud-Gaillard, J.; et al. FHL1 is a major host factor for chikungunya virus infection. Nat. Cell Biol. 2019, 574, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Schnierle, B.S. A new host factor essential for chikungunya virus. Trends Microbiol. 2020, 28, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.P.; Tritsch, S.R.; Kota, K.; Chiang, C.-Y.; Dong, L.; Kenny, T.; Brueggemann, E.E.; Ward, M.D.; Cazares, L.H.; Bavari, S. Sphingosine kinase 2 is a chikungunya virus host factor co-localized with the viral replication complex. Emerg. Microbes Infect. 2015, 4, e61. [Google Scholar] [CrossRef] [PubMed]
- Tossavainen, H.; Aitio, O.; Hellman, M.; Saksela, K.; Permi, P. Structural basis of the high affinity interaction between the alphavirus nonstructural protein-3 (nsP3) and the SH3 domain of amphiphysin-2. J. Biol. Chem. 2016, 291, 16307–16317. [Google Scholar] [CrossRef]
- Michie, A.; Ernst, T.; Chua, I.-L.J.; Lindsay, M.D.A.; Neville, P.J.; Nicholson, J.; Jardine, A.; MacKenzie, J.S.; Smith, D.W.; Imrie, A. Phylogenetic and timescale analysis of Barmah forest virus as inferred from genome sequence analysis. Viruses 2020, 12, 732. [Google Scholar] [CrossRef]
- Prokic, I.; Cowling, B.S.; Laporte, J. Amphiphysin 2 (BIN1) in physiology and diseases. J. Mol. Med. 2014, 92, 453–463. [Google Scholar] [CrossRef]
- Mim, C.; Unger, V.M. Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 2012, 37, 526–533. [Google Scholar] [CrossRef]
- Kristensen, O. Crystal structure of the G3BP2 NTF2-like domain in complex with a canonical FGDF motif peptide. Biochem. Biophys. Res. Commun. 2015, 467, 53–57. [Google Scholar] [CrossRef]
- Scholte, F.; Tas, A.; Albulescu, I.C.; Žusinaite, E.; Merits, A.; Snijder, E.J.; Van Hemert, M.J. Stress granule components G3BP1 and G3BP2 play a proviral role early in chikungunya virus replication. J. Virol. 2015, 89, 4457–4469. [Google Scholar] [CrossRef]
- Vasiljeva, L.; Merits, A.; Auvinen, P.; Kaariainen, L. Identification of a novel function of the alphavirus capping apparatus. RNA 5′-triphosphatase activity of Nsp2. J. Biol. Chem. 2000, 275, 17281–17287. [Google Scholar] [CrossRef]
- Lukash, T.; Agback, T.; Dominguez, F.; Shiliaev, N.; Meshram, C.; Frolova, E.I.; Agback, P.; Frolov, I. Structural and functional characterization of host FHL1 protein interaction with hypervariable domain of chikungunya virus nsP3 protein. J. Virol. 2020, 95, e01672-20. [Google Scholar] [CrossRef]
- Lopez, S.; Bell, J.R.; Strauss, E.G.; Strauss, J.H. The nonstructural proteins of Sindbis virus as studied with an antibody specific for the C terminus of the nonstructural readthrough polyprotein. Virology 1985, 141, 235–247. [Google Scholar] [CrossRef]
- Strauss, E.G.; Levinson, R.; Rice, C.M.; Dalrymple, J.; Strauss, J.H. Nonstructural proteins nsP3 and nsP4 of Ross River and O’Nyong-nyong viruses: Sequence and comparison with those of other alphaviruses. Virology 1988, 164, 265–274. [Google Scholar] [CrossRef]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [CrossRef]
- De Groot, R.J.; Rumenapf, T.; Kuhn, R.J.; Strauss, E.G.; Strauss, J.H. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Proc. Natl. Acad. Sci. USA 1991, 88, 8967–8971. [Google Scholar] [CrossRef]
- Merits, A.; Vasiljeva, L.; Ahola, T.; Kääriäinen, L.; Auvinen, P. Proteolytic processing of Semliki forest virus-specific non-structural polyprotein by nsP2 protease. J. Gen. Virol. 2001, 82, 765–773. [Google Scholar] [CrossRef]
- Takkinen, K.; Peranen, J.; Kaariainen, L. Proteolytic processing of Semliki forest virus-specific non-structural polyprotein. J. Gen. Virol. 1991, 72, 1627–1633. [Google Scholar] [CrossRef]
- Li, G.; Rice, C.M. The signal for translational readthrough of a UGA codon in Sindbis virus RNA involves a single cytidine residue immediately downstream of the termination codon. J. Virol. 1993, 67, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.-S.; Ng, M.M.-L.; Chu, J.J.H. Replication of alphaviruses: A review on the entry process of alphaviruses into cells. Adv. Virol. 2011, 2011, 249640. [Google Scholar] [CrossRef]
- Takkinen, K. Complete nucleotide sequence of the nonstructural protein genes of Semliki Forest virus. Nucleic Acids Res. 1986, 14, 5667–5682. [Google Scholar] [CrossRef][Green Version]
- Teo, T.-H.; Her, Z.; Tan, J.J.L.; Lum, F.-M.; Lee, W.W.L.; Chan, Y.-H.; Ong, R.-Y.; Kam, Y.-W.; Leparc-Goffart, I.; Gallian, P.; et al. Caribbean and La Réunion Chikungunya virus isolates differ in their capacity to induce proinflammatory Th1 and NK cell responses and acute joint pathology. J. Virol. 2015, 89, 7955–7969. [Google Scholar] [CrossRef] [PubMed]
- Stapleford, K.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C.; et al. Whole-genome sequencing analysis from the Chikungunya virus Caribbean outbreak reveals novel evolutionary genomic elements. PLoS Negl. Trop. Dis. 2016, 10, e0004402. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Kam, Y.-W.; Lin, R.T.P.; Ng, M.M.-L.; Ng, L.F.; Chu, J.J.H. Comparative analysis of the genome sequences and replication profiles of chikungunya virus isolates within the East, Central and South African (ECSA) lineage. Virol. J. 2013, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Tricou, V.; Selekon, B.; Faye, O.; Gessain, A.; Kazanji, M.; Nakouné, E.; Berthet, N. Complete genome sequences of Igbo-Ora and Babanki alphavirus strains isolated in the Central African Republic in the 1960s and 1970s. Microbiol. Resour. Announc. 2019, 8, e00868-19. [Google Scholar] [CrossRef]
- Simpson, D.A.; Davis, N.L.; Lin, S.-C.; Russell, D.; Johnston, R.E. Complete nucleotide sequence and full-length cDNA clone of SA AR86, a South African alphavirus related to Sindbis. Virology 1996, 222, 464–469. [Google Scholar] [CrossRef]
- Li, G.P.; Rice, C.M. Mutagenesis of the in-frame opal termination codon preceding nsP4 of Sindbis virus: Studies of translational readthrough and its effect on virus replication. J. Virol. 1989, 63, 1326–1337. [Google Scholar] [CrossRef]
- Warmbrod, K.L.; Patterson, E.I.; Kautz, T.F.; Stanton, A.; Rockx-Brouwer, D.; Kalveram, B.K.; Khanipov, K.; Thangamani, S.; Fofanov, Y.; Forrester, N.L. Viral RNA-dependent RNA polymerase mutants display an altered mutation spectrum resulting in attenuation in both mosquito and vertebrate hosts. PLoS Pathog. 2019, 15, e1007610. [Google Scholar] [CrossRef]
- Ross, R.W. The Newala epidemic: III. The virus: Isolation, pathogenic properties and relationship to the epidemic. J. Hyg. 1956, 54, 177–191. [Google Scholar] [CrossRef]
- Coffey, L.L.; Failloux, A.-B.; Weaver, S.C. Chikungunya virus-vector interactions. Viruses 2014, 6, 4628–4663. [Google Scholar] [CrossRef]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.-C.; Lavenir, R.; Pardigon, N.; Reynes, J.-M.; Pettinelli, F.; et al. Genome microevolution of Chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006, 3, e263. [Google Scholar] [CrossRef]
- Jupp, P.; McIntosh, B. Chikungunya Virus Disease. In The Arboviruses: Epidemiology and Ecology; CRC Press: Boca Raton, FL, USA, 1988; Volume 2, p. 137. [Google Scholar]
- Powers, A.M.; Logue, C.H. Changing patterns of chikungunya virus: Re-emergence of a zoonotic arbovirus. J. Gen. Virol. 2007, 88, 2363–2377. [Google Scholar] [CrossRef]
- Thiberville, S.-D.; Moyen, N.; Dupuis-Maguiraga, L.; Nougairede, A.; Gould, E.A.; Roques, P.; de Lamballerie, X. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antivir. Res. 2013, 99, 345–370. [Google Scholar] [CrossRef]
- De Lamballerie, X.; Leroy, E.; Charrel, R.N.; Ttsetsarkin, K.; Higgs, S.; Gould, E.A. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: A sign of things to come? Virol. J. 2008, 5, 33–34. [Google Scholar] [CrossRef]
- Abubakar, S.; Sam, I.-C.; Wong, P.-F.; Hooi, P.-S.; Roslan, N.; MatRahim, N. Reemergence of endemic Chikungunya, Malaysia. Emerg. Infect. Dis. 2007, 13, 147–149. [Google Scholar] [CrossRef]
- Sam, I.-C.; Chan, Y.F.; Chan, S.Y.; Loong, S.K.; Chin, H.K.; Hooi, P.S.; Ganeswrie, R.; AbuBakar, S. Chikungunya virus of Asian and Central/East African genotypes in Malaysia. J. Clin. Virol. 2009, 46, 180–183. [Google Scholar] [CrossRef]
- Hammon, W.M.; Rundnick, A.; Sather, G.E. Viruses associated with epidemic hemorrhagic fevers of the Philippines and Thailand. Science 1960, 131, 1102–1103. [Google Scholar] [CrossRef]
- Laras, K.; Sukri, N.C.; Larasati, R.P.; Bangs, M.J.; Kosim, R.; Wandra, T.; Master, J.; Kosasih, H.; Hartati, S.; Beckett, C.; et al. Tracking the re-emergence of epidemic chikungunya virus in Indonesia. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 128–141. [Google Scholar] [CrossRef]
- Sam, I.; AbuBakar, S. Chikungunya virus infection. Med. J. Malays. 2006, 61, 264. [Google Scholar]
- Kumarasamy, V.; Prathapa, S.; Zuridah, H.; Chem, Y.K.; Norizah, I.; Chua, K.B. Re-emergence of Chikungunya virus in Malaysia. Med. J. Malays. 2006, 61, 221. [Google Scholar]
- Lam, S.K.; Chua, K.B.; Hooi, P.S.; Rahimah, M.A.; Kumari, S.; Tharmaratnam, M.; Chuah, S.K.; Smith, D.W.; Sampson, I.A. Chikungunya infection—An emerging disease in Malaysia. Southeast Asian J. Trop. Med. Public Health 2001, 32, 447–451. [Google Scholar]
- Ng, L.-C.; Tan, L.-K.; Tan, C.-H.; Tan, S.S.; Hapuarachchi, H.C.; Pok, K.-Y.; Lai, Y.-L.; Lam-Phua, S.-G.; Bucht, G.; Lin, R.T.; et al. Entomologic and virologic investigation of Chikungunya, Singapore. Emerg. Infect. Dis. 2009, 15, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Hapuarachchi, H.C.; Bandara, K.B.A.T.; Sumanadasa, S.D.M.; Hapugoda, M.D.; Lai, Y.-L.; Lee, K.-S.; Tan, L.-K.; Lin, R.T.P.; Ng, L.F.P.; Bucht, G.; et al. Re-emergence of Chikungunya virus in Southeast Asia: Virological evidence from Sri Lanka and Singapore. J. Gen. Virol. 2009, 91, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.-K.; Sy, A.K.D.; Tandoc, A.O.; Khoo, J.-J.; Sulaiman, S.; Chang, L.-Y.; Abubakar, S. Independent emergence of the Cosmopolitan Asian Chikungunya virus, Philippines 2012. Sci. Rep. 2015, 5, 12279. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive genome scale phylogenetic study provides new insights on the global expansion of Chikungunya Virus. J. Virol. 2016, 90, 10600–10611. [Google Scholar] [CrossRef] [PubMed]
- Cassadou, S.; Boucau, S.; Petit-Sinturel, M.; Huc, P.; Leparc-Goffart, I.; Ledrans, M. Emergence of chikungunya fever on the French side of Saint Martin Island, October to December 2013. Eurosurveillance 2014, 19, 20752. [Google Scholar] [CrossRef] [PubMed]
- Van Bortel, W.; Dorleans, F.; Rosine, J.; Blateau, A.; Rousset, D.; Matheus, S.; Leparc-Goffart, I.; Flusin, O.; Prat, C.M.; Césaire, R.; et al. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Eurosurveillance 2014, 19, 20759. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Valadere, A.M. Transcontinental movement of asian genotype Chikungunya Virus. Emerg. Infect. Dis. 2014, 20, 1400–1402. [Google Scholar] [CrossRef]
- WHO. Number of Reported Cases of Chikungunya Fever in the Americas, by Country or Territory 2015—Cumulative Cases; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Langsjoen, R.M.; Haller, S.L.; Roy, C.J.; Vinet-Oliphant, H.; Bergren, N.A.; Erasmus, J.H.; Livengood, J.A.; Powell, T.D.; Weaver, S.C.; Rossi, S.L. Chikungunya virus strains show lineage-specific variations in virulence and cross-protective ability in murine and nonhuman primate models. mBio 2018, 9, e02449-17. [Google Scholar] [CrossRef]
- Paquet, C.; Quatresous, I.; Solet, J.; Sissoko, D.; Renault, P.; Pierre, V.; Cordel, H.; Lassalle, C.; Thiria, J.; Zeller, H. Chikungunya outbreak in Reunion: Epidemiology and surveillance, 2005 to early January 2006. Weekly releases (1997–2007). Eurosurveillance 2006, 11, 2891. [Google Scholar]
- Duong, V.; Andries, A.-C.; Ngan, C.; Sok, T.; Richner, B.; Asgari-Jirhandeh, N.; Bjorge, S.; Huy, R.; Ly, S.; Laurent, D.; et al. Reemergence of Chikungunya virus in Cambodia. Emerg. Infect. Dis. 2012, 18, 2066–2069. [Google Scholar] [CrossRef]
- Pialoux, G.; Gaüzère, B.-A.; Jauréguiberry, S.; Strobel, M. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 2007, 7, 319–327. [Google Scholar] [CrossRef]
- Sahadeo, N.S.D.; Allicock, O.M.; De Salazar, P.M.; Auguste, A.; Widen, S.; Olowokure, B.; Gutierrez, C.; Valadere, A.M.; Polson-Edwards, K.; Weaver, S.C.; et al. Understanding the evolution and spread of chikungunya virus in the Americas using complete genome sequences. Virus Evol. 2017, 3, vex010. [Google Scholar] [CrossRef] [PubMed]
- Arankalle, V.A.; Shrivastava, S.; Cherian, S.; Gunjikar, R.S.; Walimbe, A.M.; Jadhav, S.M.; Sudeep, A.B.; Mishra, A.C. Genetic divergence of Chikungunya viruses in India (1963–2006) with special reference to the 2005–2006 explosive epidemic. J. Gen. Virol. 2007, 88, 1967–1976. [Google Scholar] [CrossRef]
- Sam, I.C.; Loong, S.K.; Michael, J.C.; Chua, C.L.; Sulaiman, W.Y.W.; Vythilingam, I.; Chan, S.Y.; Chiam, C.W.; Yeong, Y.S.; AbuBakar, S.; et al. Genotypic and phenotypic characterization of Chikungunya virus of different genotypes from Malaysia. PLoS ONE 2012, 7, e50476. [Google Scholar] [CrossRef]
- Ooi, M.K.; Gan, H.M.; Rohani, A.; Hassan, S.S. First complete genome sequence of a chikungunya virus strain isolated from a patient diagnosed with dengue virus infection in Malaysia. Genome Announc. 2016, 4, e00876-16. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-H.; Yang, C.-F.; Su, C.-L.; Chang, S.-F.; Cheng, C.-H.; Yu, S.-K.; Lin, C.-C.; Shu, P.-Y. Imported Chikungunya virus strains, Taiwan, 2006–2009. Emerg. Infect. Dis. 2009, 15, 1854–1856. [Google Scholar] [CrossRef]
- Leparc-Goffart, I.; Nougairede, A.; Cassadou, S.; Prat, C.; de Lamballerie, X. Chikungunya in the Americas. Lancet 2014, 383, 514. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Y.; Wang, M.; Yang, F.; Wu, C.; Huang, D.; Xiong, L.; Wan, C.; Cheng, J.; Zhang, R. Differences in genome characters and cell tropisms between two chikungunya isolates of Asian lineage and Indian Ocean lineage. Virol. J. 2018, 15, 130. [Google Scholar] [CrossRef]
- Jain, J.; Mathur, K.; Shrinet, J.; Bhatnagar, R.K.; Sunil, S. Analysis of coevolution in nonstructural proteins of chikungunya virus. Virol. J. 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Brès, V.; Yu, G.; Krysztof, D.; Naccache, S.N.; Lee, D.; Pfeil, J.; Linnen, J.M.; Stramer, S.L. Genomic assays for identification of chikungunya virus in blood donors, Puerto Rico, 2014. Emerg. Infect. Dis. 2015, 21, 1409–1413. [Google Scholar] [CrossRef]
- Savage, H.M.; Ledermann, J.P.; Yug, L.; Burkhalter, K.L.; Marfel, M.; Hancock, W.T. Incrimination of Aedes (Stegomyia) hensilli Farner as an epidemic vector of chikungunya virus on Yap Island, Federated States of Micronesia, 2013. Am. J. Trop. Med. Hyg. 2015, 92, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, S.; Vollmar, P.; Poluda, D.; Zange, S.; Antwerpen, M.H.; Löscher, T.; Dobler, G. Complete genome sequence of a chikungunya virus imported from Bali to Germany. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Sasmono, R.T.; Perkasa, A.; Yohan, B.; Haryanto, S.; Yudhaputri, F.A.; Hayati, R.F.; Ma’roef, C.N.; Ledermann, J.P.; Myint, K.S.A.; Powers, A.M. Chikungunya detection during dengue outbreak in Sumatra, Indonesia: Clinical mani-festations and virological profile. Am. J. Trop. Med. Hyg. 2017, 97, 1393–1398. [Google Scholar] [CrossRef]
- Dupont-Rouzeyrol, M.; Caro, V.; Guillaumot, L.; Vazeille, M.; D’Ortenzio, E.; Thiberge, J.-M.; Baroux, N.; Gourinat, A.-C.; Grandadam, M.; Failloux, A.-B. Chikungunya virus and the mosquito vector Aedes aegypti in New Caledonia (South Pacific Region). Vector-Borne Zoonotic Dis. 2012, 12, 1036–1041. [Google Scholar] [CrossRef]
- Sun, Y.; Yan, J.; Mao, H.; Zhang, L.; Lyu, Q.; Wu, Z.; Zheng, W.; Feng, C.; Zhang, Y. Characterization of the complete genome of Chikungunya in Zhejiang, China, using a modified virus discovery method based on cDNA-AFLP. PLoS ONE 2013, 8, e83014. [Google Scholar] [CrossRef]
- Nunes, M.R.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; de Oliveira, L.F.; da Silva Azevedo, R.D.; da Silva, D.E.; da Silva, E.V.; da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 1–11. [Google Scholar] [CrossRef]
- Suhana, O.; Nazni, W.; Apandi, Y.; Farah, H.; Lee, H.; Sofian-Azirun, M. Insight into the origin of chikungunya virus in Malaysian non-human primates via sequence analysis. Heliyon 2019, 5, e02682. [Google Scholar] [CrossRef]
- Kumar, C.P.; John, B.A.; Khna, S.A.; Lyla, P.S.; Kamaleson, A.S. Mutational patterns and DNA barcode for diagnosing Chikungunya virus. Int. J. Virol. 2011, 7, 42–52. [Google Scholar] [CrossRef][Green Version]
- Vasilakis, N.; Cardosa, J.; Diallo, M.; Sall, A.A.; Holmes, E.C.; Hanley, K.A.; Weaver, S.C.; Mota, J.; Rico-Hesse, R. Sylvatic Dengue viruses share the pathogenic potential of urban/endemic Dengue viruses. J. Virol. 2010, 84, 3726–3728. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Bordería, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef]
- Mosimann, A.L.P.; De Siqueira, M.K.; Ceole, L.F.; Dos Santos, C.N.D. A new Aura virus isolate in Brazil shows segment duplication in the variable region of the nsP3 gene. Parasites Vectors 2018, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; Da Rosa, A.P.T.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, W.I.; Tesh, R.B.; et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Brault, A.C.; Kang, W.; Holland, J.J. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 1999, 73, 4316–4326. [Google Scholar] [CrossRef] [PubMed]
- Shirako, Y.; Niklasson, B.; Dalrymple, J.M.; Strauss, E.G.; Strauss, J.H. Structure of the Ockelbo virus genome and its relationship to other Sindbis viruses. Virology 1991, 182, 753–764. [Google Scholar] [CrossRef]
- Weinbren, M.P.; Kokernot, R.H.; Smithburn, K.C. Strains of Sindbis-like virus isolated from culicine mosquitoes in the Union of South Africa. I. Isolation and properties. S. Afr. Med. J. 1956, 30, 631–636. [Google Scholar]
- Malherbe, H.; Strickland-Cholmley, M.; Jackson, A.L. Sindbis virus infection in man. Report of a case with recovery of virus from skin lesions. S. Afr. Med. J. 1963, 37, 547–552. [Google Scholar]
- Pickering, P.; Aaskov, J.G.; Liu, W. Complete genomic sequence of an Australian Sindbis virus isolated 44 years ago reveals unique indels in the E2 and nsP3 proteins. Microbiol. Resour. Announc. 2019, 8, e00246-19. [Google Scholar] [CrossRef]
- Smithburn, K.C.; Haddow, A. Semliki forest virus. I. Isolation and pathogenic properties. J. Immunol. 1944, 49, 141–157. [Google Scholar]
- Bradish, C.J.; Allner, K.; Maber, H.B. The virulence of original and derived strains of Semliki forest virus for mice, guinea-pigs and rabbits. J. Gen. Virol. 1971, 12, 141–160. [Google Scholar] [CrossRef]
- Amor, S.; Scallan, M.F.; Morris, M.M.; Dyson, H.; Fazakerley, J.K. Role of immune responses in protection and pathogenesis during Semliki Forest virus encephalitis. J. Gen. Virol. 1996, 77, 281–291. [Google Scholar] [CrossRef]
- Fazakerley, J.; Pathak, S.; Scallan, M.; Amor, S.; Dyson, H. Replication of the A7(74) strain of Semliki forest virus is restricted in neurons. Virology 1993, 195, 627–637. [Google Scholar] [CrossRef]
- Oliver, K.R.; Scallan, M.F.; Dyson, H.; Fazakerley, J.K. Susceptibility to a neurotropic virus and its changing distribution in the developing brain is a function of CNS maturity. J. Neurovirol. 1997, 3, 38–48. [Google Scholar] [CrossRef]
- Glasgow, G.M.; Sheahan, B.J.; Atkins, G.J.; Wahlberg, J.M.; Salminen, A.; Lilieström, P. Two mutations in the envelope glycoprotein E2 of Semliki forest virus affecting the maturation and entry patterns of the virus alter pathogenicity for mice. Virology 1991, 185, 741–748. [Google Scholar] [CrossRef]
- Atkins, G.J.; Sheahan, B.J.; Mooney, D.A. Pathogenicity of Semliki Forest virus for the rat central nervous system and primary rat neural cell cultures: Possible implications for the pathogenesis of multiple sclerosis. Neuropathol. Appl. Neurobiol. 1990, 16, 57–68. [Google Scholar] [CrossRef]
- Peränen, J.; Kääriäinen, L. Biogenesis of type I cytopathic vacuoles in Semliki Forest virus-infected BHK cells. J. Virol. 1991, 65, 1623–1627. [Google Scholar] [CrossRef]
- Peranen, J. Localization and phosphorylation of Semliki forest virus non-structural protein nsP3 expressed in COS cells from a cloned cDNA. J. Gen. Virol. 1991, 72, 195–199. [Google Scholar] [CrossRef]
- Lau, C.; Aubry, M.; Musso, D.; Teissier, A.; Paulous, S.; Desprès, P.; De-Lamballerie, X.; Pastorino, B.; Cao-Lormeau, V.-M.; Weinstein, P. New evidence for endemic circulation of Ross River virus in the Pacific Islands and the potential for emergence. Int. J. Infect. Dis. 2017, 57, 73–76. [Google Scholar] [CrossRef]
- Rosen, L.; Gubler, D.J.; Bennett, P.H. Epidemic polyarthritis (Ross River) virus infection in the Cook Islands. Am. J. Trop. Med. Hyg. 1981, 30, 1294–1302. [Google Scholar] [CrossRef]
- Sammels, L.M.; Coelen, R.J.; Lindsay, M.D.; MacKenzie, J.S. Geographic distribution and evolution of Ross River Virus in Australia and the Pacific Islands. Virology 1995, 212, 20–29. [Google Scholar] [CrossRef][Green Version]
- Claflin, S.B.; Webb, C.E. Ross River virus: Many vectors and unusual hosts make for an unpredictable pathogen. PLoS Pathog. 2015, 11, e1005070. [Google Scholar] [CrossRef]
- Russell, R.C. Ross River virus: Ecology and distribution. Annu. Rev. Entomol. 2002, 47, 1–31. [Google Scholar] [CrossRef]
- Bielefeldt-Ohmann, H.; Barclay, J. Pathogenesis of Ross River virus-induced diseases: A role for viral quasispecies and persistence. Microb. Pathog. 1998, 24, 373–383. [Google Scholar] [CrossRef]
- Smith, D.W.; Speers, D.J.; MacKenzie, J.S. The viruses of Australia and the risk to tourists. Travel Med. Infect. Dis. 2011, 9, 113–125. [Google Scholar] [CrossRef]
- Doherty, R.; Carley, J.; Kay, B.; Filippich, C.; Marks, E.N.; Frazier, C.L. Isolation of virus strains from mosquitoes collected in Queensland, 1972–1976. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 509–520. [Google Scholar] [CrossRef]
- Marshall, I.D.; Woodroofe, G.M.; Hirsch, S. Viruses recovered from mosquitoes and wildlife serum collected in the Murray Valley of South-Eastern Australia, February 1974, during an epidemic of encephalitis. Aust. J. Exp. Biol. Med. Sci. 1982, 60, 457–470. [Google Scholar] [CrossRef]
- Boughton, C.R.; Hawkes, R.A.; Nairn, H.M. Illness caused by a Barmah Forest-like virus in New South Wales. Med. J. Aust. 1988, 148, 146–147. [Google Scholar] [CrossRef]
- Merianos, A.; Farland, A.; Patel, M.; Currie, B.; Whelan, P.; Dentith, H.; Smith, D. A concurrent outbreak of Barmah Forest and Ross River virus disease in Nhulunbuy, Northern Territory. Commun. Dis. Intell. 1992, 16, 110–111. [Google Scholar]
- Lindsay, M.D.; Johansen, C.A.; Broom, A.K.; Smith, D.W.; MacKenzie, J.S. Emergence of Barmah Forest Virus in Western Australia. Emerg. Infect. Dis. 1995, 1, 22–26. [Google Scholar] [CrossRef]
- Wishart, E.; O’Grady, K.A.; Passmore, J.; Moran, R. An outbreak of Barmah Forest virus disease in Victoria. Commun. Dis. Intell. Q. Rep. 2002, 26, 600. [Google Scholar]
- Caly, L.; Horwood, P.F.; Vijaykrishna, D.; Lynch, S.; Greenhill, A.R.; Pomat, W.; Rai, G.; Kisa, D.; Bande, G.; Druce, J.; et al. Divergent Barmah forest virus from Papua New Guinea. Emerg. Infect. Dis. 2019, 25, 2266–2269. [Google Scholar] [CrossRef]
- Morley, V.J.; Noval, M.G.; Chen, R.; Weaver, S.C.; Vignuzzi, M.; Stapleford, K.A.; Turner, P.E. Chikungunya virus evolution following a large 3′UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol. 2018, 4, vey012. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C. Mosquito-borne arboviruses in Australia: The current scene and implications of climate change for human health. Int. J. Parasitol. 1998, 28, 955–969. [Google Scholar] [CrossRef]
- Ehlkes, L.; Eastwood, K.; Webb, C.; Durrheim, D. Surveillance should be strengthened to improve epidemiological understandings of mosquito-borne Barmah Forest virus infection. West. Pac. Surveill. Response J. 2012, 3, 63. [Google Scholar] [CrossRef]
- Nagy, P.D.; Simon, A.E. New insights into the mechanisms of RNA recombination. Virology 1997, 235, 1–9. [Google Scholar] [CrossRef]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.E. In vitro synthesis of infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Oberste, M.; Parker, M.D.; Smith, J.F. Complete sequence of Venezuelan Equine Encephalitis virus subtype IE reveals conserved and hypervariable domains within the C terminus of nsP3. Virology 1996, 219, 314–320. [Google Scholar] [CrossRef]
- Meissner, J.D.; Huang, C.Y.-H.; Pfeffer, M.; Kinney, R.M. Sequencing of prototype viruses in the Venezuelan equine encephalitis antigenic complex. Virus Res. 1999, 64, 43–59. [Google Scholar] [CrossRef]
- Kallio, K.; Hellström, K.; Balistreri, G.; Spuul, P.; Jokitalo, E.; Ahola, T. Template RNA length determines the size of replication complex spherules for Semliki forest virus. J. Virol. 2013, 87, 9125–9134. [Google Scholar] [CrossRef]
- Rupp, J.C.; Jundt, N.; Hardy, R.W. Requirement for the amino-terminal domain of Sindbis virus nsP4 during virus infection. J. Virol. 2011, 85, 3449–3460. [Google Scholar] [CrossRef]
- Keck, F.; Ataey, P.; Amaya, M.; Bailey, C.; Narayanan, A. Phosphorylation of single stranded RNA virus proteins and potential for novel therapeutic strategies. Viruses 2015, 7, 5257–5273. [Google Scholar] [CrossRef]
- Peranen, J.; Takkinen, K.; Kalkkinen, N.; Kaariainen, L. Semliki forest virus-specific non-structural protein nsP3 is a phosphoprotein. J. Gen. Virol. 1988, 69, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; La Starza, M.W.; Hardy, W.R.; Strauss, J.H.; Rice, C.M. Phosphorylation of Sindbis virus nsP3 in vivo and in vitro. Virology 1990, 179, 416–427. [Google Scholar] [CrossRef]
- Vihinen, H.; Saarinen, J. Phosphorylation site analysis of Semliki forest virus nonstructural protein 3. J. Biol. Chem. 2000, 275, 27775–27783. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; De La Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of IKKβ in Venezuelan Equine Encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef]
- LaStarza, M.W.; Grakoui, A.; Rice, C.M. Deletion and duplication mutations in the C-terminal nonconserved region of Sindbis virus nsP3: Effects on phosphorylation and on virus replication in vertebrate and invertebrate cells. Virology 1994, 202, 224–232. [Google Scholar] [CrossRef]
- Barton, D.J.; Sawicki, S.G.; Sawicki, D.L. Solubilization and immunoprecipitation of alphavirus replication complexes. J. Virol. 1991, 65, 1496–1506. [Google Scholar] [CrossRef]
- Bara, J.J.; Muturi, E.J. Effect of mixed infections of Sindbis and La Crosse viruses on replication of each virus in vitro. Acta Trop. 2014, 130, 71–75. [Google Scholar] [CrossRef]
- Newman, C.M.; Cerutti, F.; Anderson, T.K.; Hamer, G.L.; Walker, E.D.; Kitron, U.D.; Ruiz, M.O.; Brawn, J.D.; Goldberg, T.L. Culex Flavivirus and West Nile virus mosquito coinfection and positive ecological association in Chicago, United States. Vector-Borne Zoonotic Dis. 2011, 11, 1099–1105. [Google Scholar] [CrossRef]
- Kent, R.J.; Crabtree, M.B.; Miller, B.R. Transmission of West Nile virus by Culex quinquefasciatus Say infected with Culex Flavivirus Izabal. PLoS Negl. Trop. Dis. 2010, 4, e671. [Google Scholar] [CrossRef]
- Pepin, K.M.; Hanley, K.A. Density-dependent competitive suppression of Sylvatic Dengue Virus by Endemic Dengue Virus in cultured mosquito cells. Vector-Borne Zoonotic Dis. 2008, 8, 821–828. [Google Scholar] [CrossRef]
- Pepin, K.M.; Lambeth, K.; Hanley, K.A. Asymmetric competitive suppression between strains of dengue virus. BMC Microbiol. 2008, 8, 28. [Google Scholar] [CrossRef]
- Potiwat, R.; Komalamisra, N.; Thavara, U.; Tawatsin, A.; Siriyasatien, P. Competitive suppression between chikungunya and dengue virus in Aedes albopictus c6/36 cell line. Southeast Asian J. Trop. Med. Public. Health 2011, 42, 1388–1394. [Google Scholar]
- Mosquera, J.; Adler, F.R. Evolution of virulence: A unified framework for coinfection and superinfection. J. Theor. Biol. 1998, 195, 293–313. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
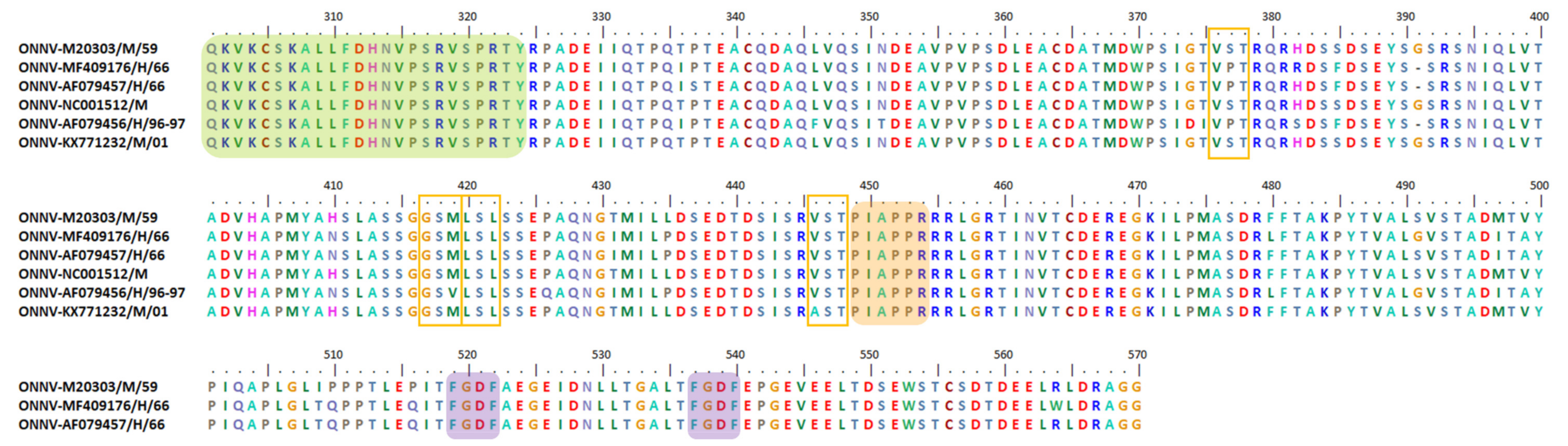{kind=link}
| 4 aa Deletion at Position 379–382 | |||
|---|---|---|---|
| Strain | GenBank Accession Number | Lineage | Details |
| Indonesia/0712aTW | FJ807886 | Asian | All samples were taken from febrile patients who arrived at two Taiwanese airports from January 2006 to February 2009. Seven strains imported from Indonesia had 4 aa deletions in their nsP3 HVD. The Indonesia/0706aTW isolate shares a 99.42% genetic identity with MY0031MR, a CHIKV human isolate from Malaysia. MY0031MR was isolated in Bagan Panchor in 2006 [239,240]. |
| Indonesia/0712bTW | FJ807887 | ||
| Indonesia/0802aTW | FJ807888 | ||
| Indonesia/0804aTW | FJ807889 | ||
| Indonesia/0806aTW | FJ807890 | ||
| Indonesia/0811aTW | FJ807891 | ||
| Indonesia/0706aTW | FJ807897 | ||
| 14.02217 | KY435477 | Asian | The 14.02217 was isolated in 2014 from Guyana during an American CHIKV outbreak. It belongs to the Asian/American lineage [235]. |
| PR-S6 | KR264951 | Asian | These four strains were isolated from South America in 2014: PR-S6 (Puerto Rico), 99,659 (British Virgin Islands), InDRE 51CHIK (Mexico), and InDRE 4CHIK (Mexico) [229,242,243]. |
| 99659 | KJ451624 | ||
| InDRE 51CHIK | KP851709 | ||
| InDRE 4CHIK | KP851710 | ||
| Yap 13–2039 | KJ689452 | Asian | These four strains were isolated from Micronesia in 2013 from different hosts. The Yap 13–2039 was isolated from the Ae. aegypti mosquito pool, the Yap 13–2148 from the Aedes hensilli mosquito pool, and both 3807 and 3462 from human samples [229,242,244]. |
| Yap 13–2148 | KJ689453 | ||
| 3807 | KJ451622 | ||
| 3462 | KJ451623 | ||
| 7 aa Deletion Events at Position 376–382 | |||
| MY/06/37348 | FN295483 | Asian | These strains are the earliest CHIKV strains isolated from Malaysia. They were isolated from a CHIKV outbreak in Bagan Panchor Perak in March 2006, which took place concurrently with the global ECSA strain outbreaks. The amplification and sequencing were conducted using original patient serum, confirming that the deletion event occurred during the outbreak of this virus strain and did not arise from laboratory passaging [216,217,237]. |
| MY/06/37350 | FN295484 | ||
| JMB-154 | KX097982 | Asian | JMB-154 was isolated during a DENV outbreak in Jambi, Indonesia, in 2015 from a patient with DENV-like illness symptoms. However, the patient was confirmed to be negative for DENV. The DH130003 was isolated from a traveler who returned to Germany after visiting Bali, Indonesia, in 2013 [241,245,246]. |
| DH130003 | KM673291 | ||
| SZ1239 | MG664851 | Asian | SZ1239 was isolated from a traveler after visiting Indonesia in 2012. It shares a 98.5% identity with the DH130003 strain [241]. |
| NC/2011–568 | HE806461 | Asian | The NC/2011–568 was isolated from the sample of the first reported human autochthonous case in New Caledonia in 2011 [242,247]. |
| chikv-sy | KF318729 | Asian | The Zhejiang/chik-sy/2012 was the first CHIKV isolate from Zhejiang, China, in 2012. Phylogenetic analysis showed that this strain has a high homology with a strain isolated in Taiwan, which was originally derived from Indonesia, the Indonesia/0706aTw/2007 strain [242,248]. |
| PER160/H803609 | KP164571 | Asian | These two strains were the first autochthonous CHIKV samples isolated from various parts of Brazil in 2014 [242,249]. |
| AMA2798/H804298 | KP164567 | ||
| 76 aa duplication event at position 376–456 | |||
| MUM01–2009-Selangor | KX168429 | Asian | The MUM01–2009-Selangor was isolated from a DENV2-positive patient sample. It shares a 99% genetic similarity with MY/06/37348 and MY/06/37350 [238]. |
| nsP3 HVD Residues | Predicted Co-Evolving Partners Residues in nsP3 HVD | Details and Suggested Functions |
|---|---|---|
| 361D: Located in fragment A. It is also present as a duplicate in the CHIKV MUM01–2009-Selangor strain motif (I) at position 437 (Figure 2b). | 408R and 411T: Both are residues located in fragment B (Figure 2a) and at position 477 (Figure 2b). | The 361D residue has 18 co-evolution partners, and 7 of them have the most significant interactions [242]. Both co-evolution interactions between 361D and 408R and 411T are suggested to affect the host cell cycle and modulate the level of proteolysis and peptidolysis activity [242] |
| 464P: Located in fragment C3 (Figure 2a) and at position 533 (Figure 2b). | 327S: Located in the N-terminal nsP3 HVD and present as a duplicate in the CHIKV MUM01–2009-Selangor strain motif (I) at position 413 (Figure 2b). | The co-evolution is suggested to affect the aa phosphorylation and serine or threonine kinase activity [248,250,251]. The co-evolution interaction is also suggested to be involved in signal transduction—for example, in TGF-β signaling and NF-κB activation [252,253]. |
| 377H: Located in fragment A and present only in CHIKV strains without the 7 aa deletion or 76 aa duplication (Figure 2a). | The 377H and 381S are present in the 4 or 7 aa deleted residues in CHIKV Asian strains. These three co-evolution interactions between 464P and 377H, 381S, and 132M are suggested to be involved in aa phosphorylation and modulate the level of serine or threonine kinase activity [242,254,255]. | |
| 381S: Located in fragment A and present only in CHIKV strains without the 4 or 7 aa deletion or 76 aa duplication (Figure 2a). | ||
| 132M: Located in the nsP3 macrodomain | ||
| 395S: Located in fragment B (Figure 2) and at position 464 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | This interaction was predicted to be involved in the modulation of cell communication, SH3-binding domain, focal adhesion, and virus signal transduction [242]. | |
| 395S: Located in fragment B (Figure 2) and at position 464 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | 463H: Located in fragment C3 (Figure 2) and at position 532 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | This interaction was predicted to be involved in the modulation of cell communication, SH3-binding domain, focal adhesion, and virus signal transduction [242]. |
| 411T: Located in fragment B (Figure 2) and at position 480 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | 455P: Located in fragment C2 (Figure 2) and at position 524 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | The interaction is suggested to be involved in receptor signaling, the regulation of circadian rhythm, the response to UV, and protein kinase CK2 activity [242]. |
| 408R: Located in fragment B (Figure 2) and at position 477 in the CHIKV MUM01–2009-Selangor strain (Figure 2b). | This interaction is suggested to be involved in glycosyl group transfer and activation of the MAPK pathway [242]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah, N.; Ahemad, N.; Aliazis, K.; Khairat, J.E.; Lee, T.C.; Abdul Ahmad, S.A.; Adnan, N.A.A.; Macha, N.O.; Hassan, S.S. The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence. Viruses 2021, 13, 1021. https://doi.org/10.3390/v13061021
Abdullah N, Ahemad N, Aliazis K, Khairat JE, Lee TC, Abdul Ahmad SA, Adnan NAA, Macha NO, Hassan SS. The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence. Viruses. 2021; 13(6):1021. https://doi.org/10.3390/v13061021
Chicago/Turabian StyleAbdullah, Nurshariza, Nafees Ahemad, Konstantinos Aliazis, Jasmine Elanie Khairat, Thong Chuan Lee, Siti Aisyah Abdul Ahmad, Nur Amelia Azreen Adnan, Nur Omar Macha, and Sharifah Syed Hassan. 2021. "The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence" Viruses 13, no. 6: 1021. https://doi.org/10.3390/v13061021
APA StyleAbdullah, N., Ahemad, N., Aliazis, K., Khairat, J. E., Lee, T. C., Abdul Ahmad, S. A., Adnan, N. A. A., Macha, N. O., & Hassan, S. S. (2021). The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence. Viruses, 13(6), 1021. https://doi.org/10.3390/v13061021