
An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.2. RNA Extraction and Next-Generation Sequencing
2.3. Phylogenetic and Sequence Analysis
3. Results
3.1. Site Selection and Clinical Analysis
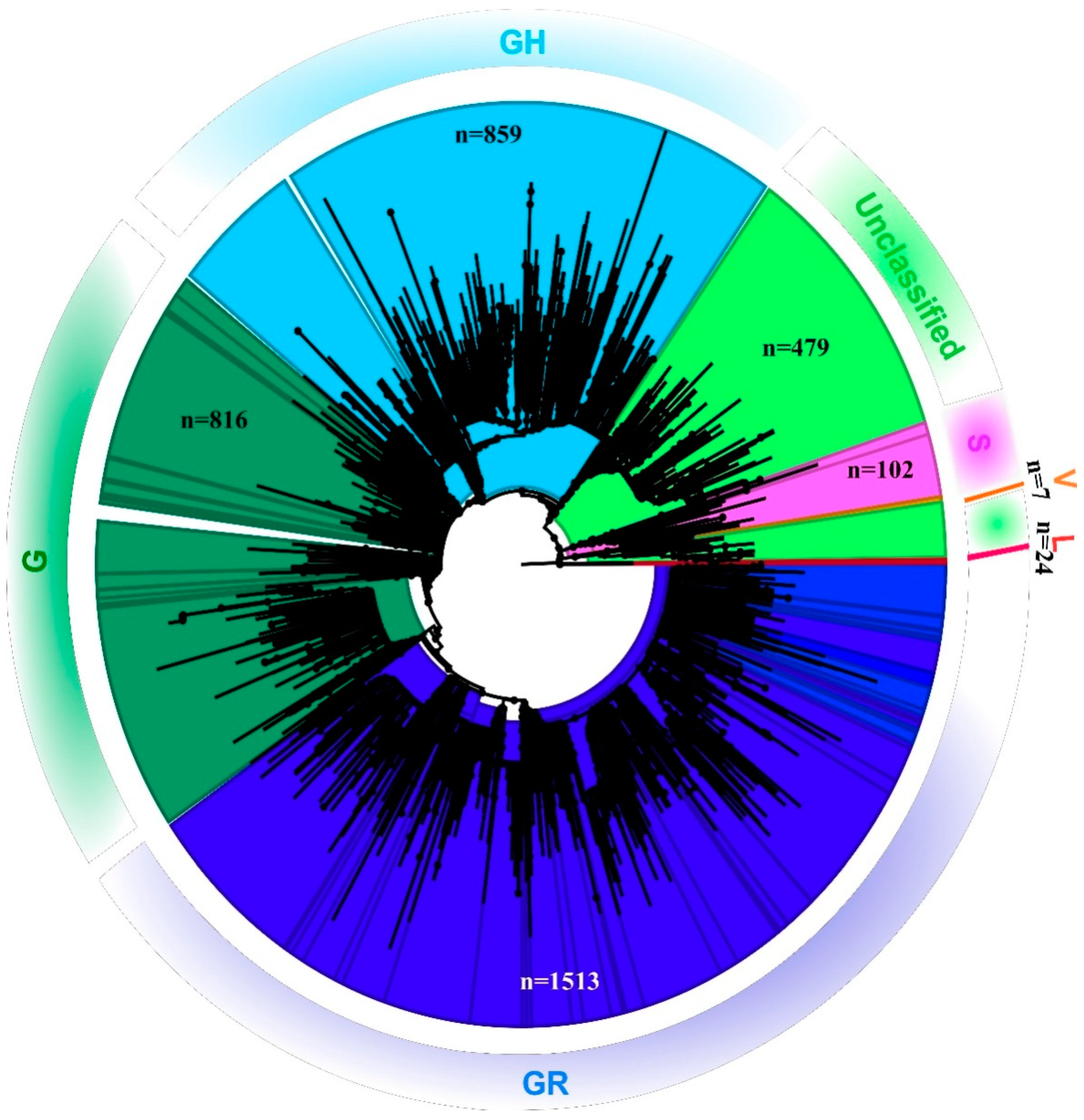
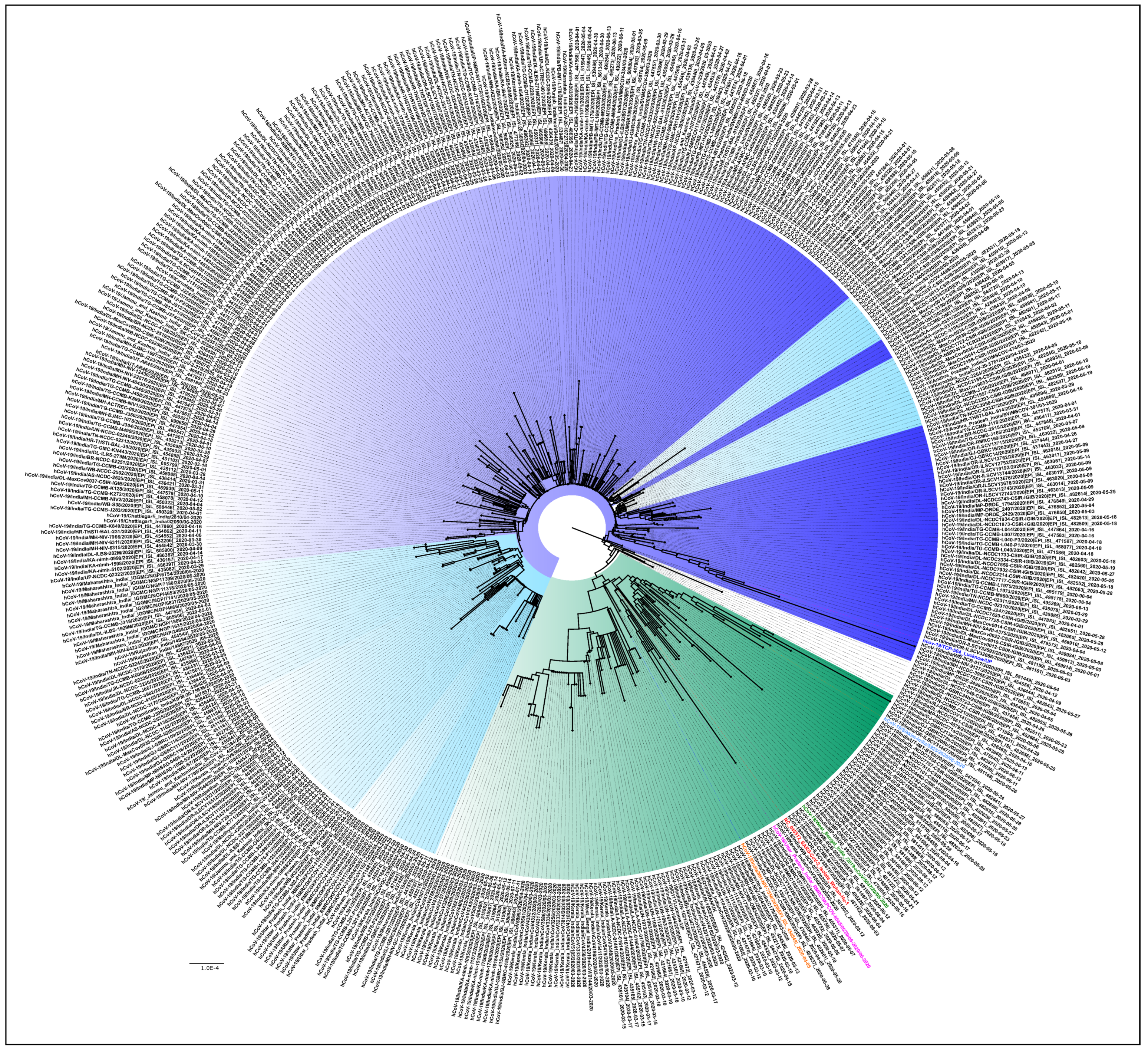
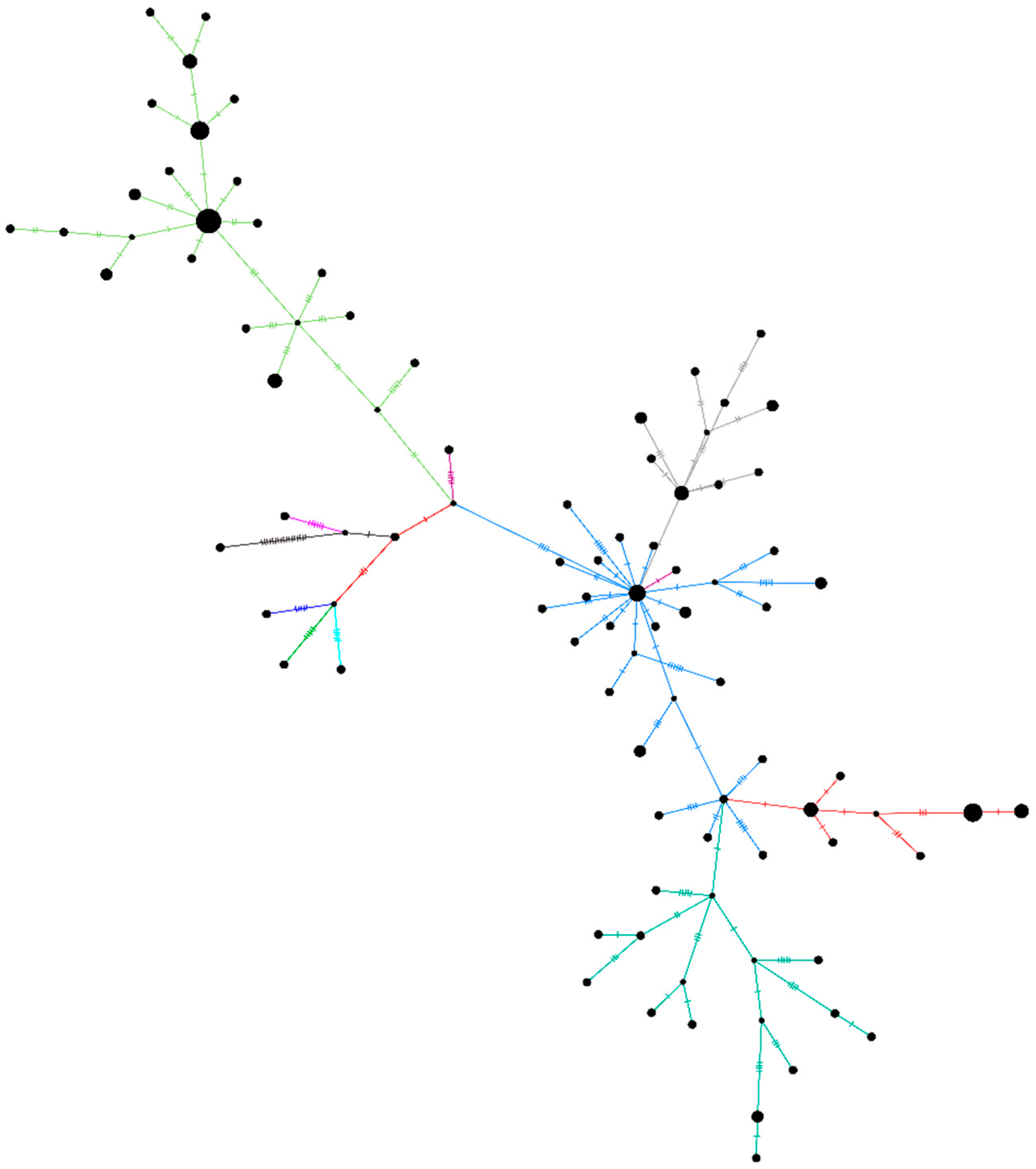
3.2. NGS Data Selection and Phylogenetic Analysis
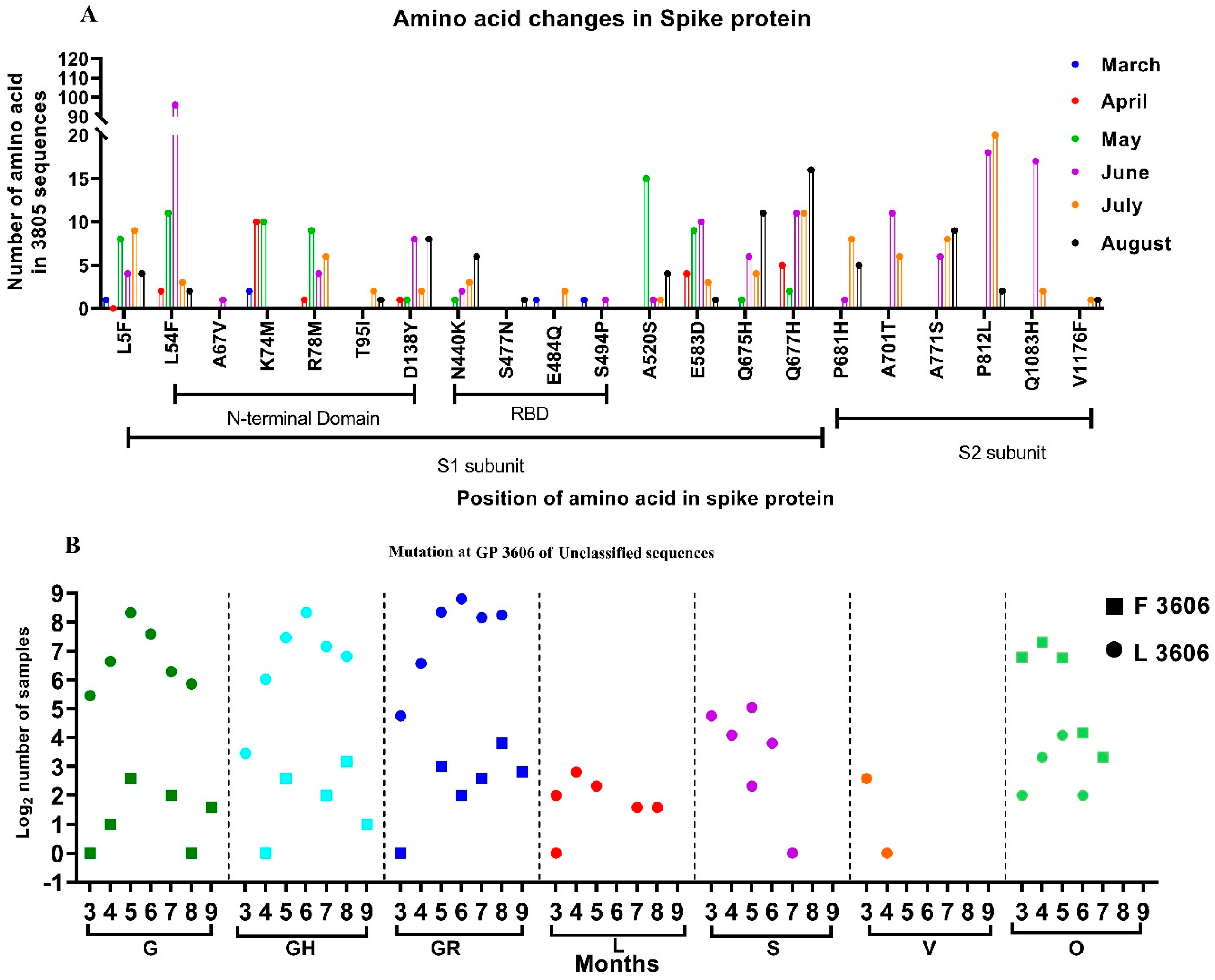
3.3. Mutations Observed in the SARS-CoV-2 Sequences
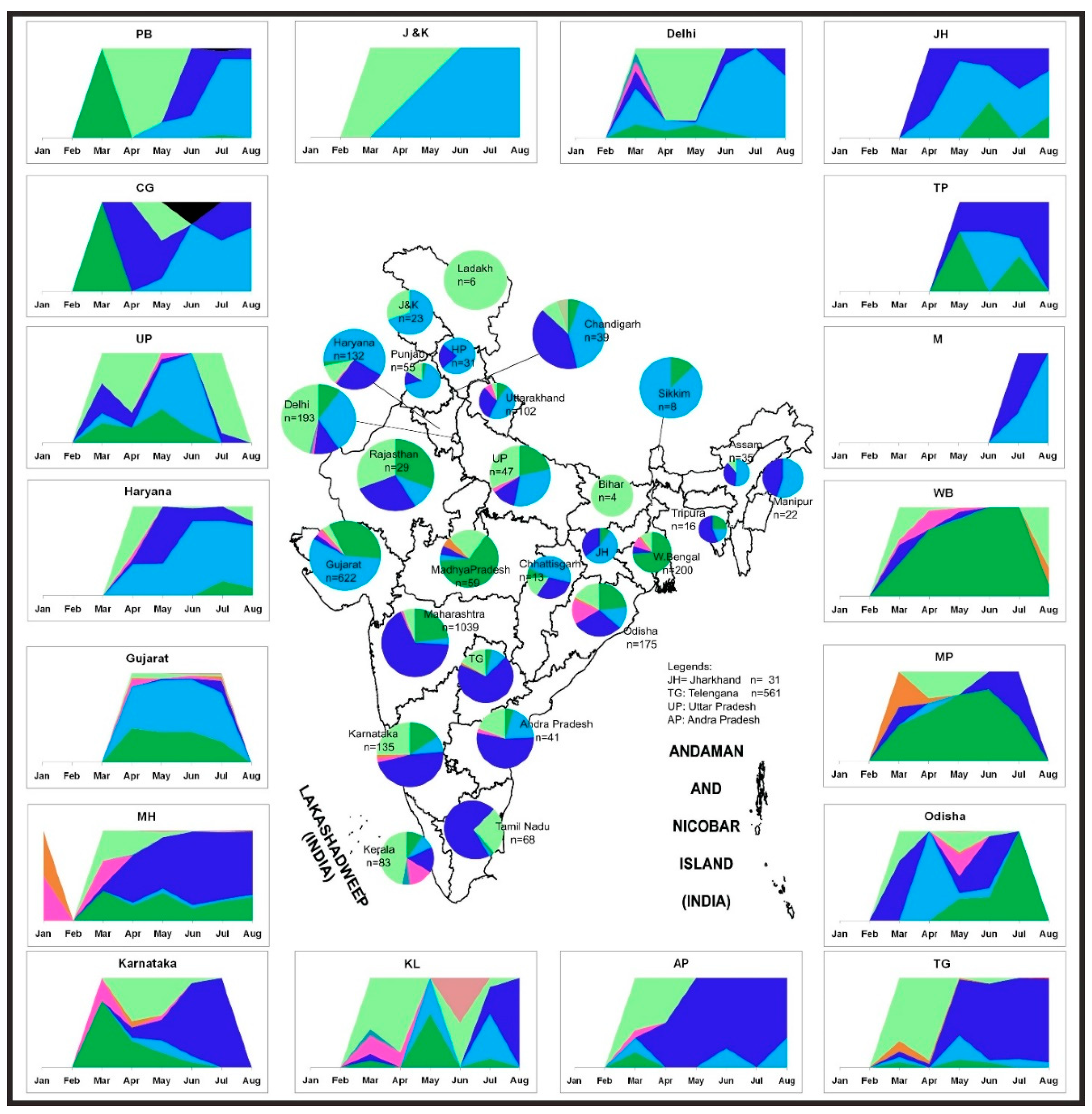
3.4. The Temporal Trend of Indian SARS-CoV-2 Sequences Demonstrates an Increase of G and Its Subclades in Different States of India
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novel Coronavirus (2019-NCoV) Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 7 April 2020).
- Yadav, P.D.; Potdar, V.A.; Choudhary, M.L.; Nyayanit, D.A.; Agrawal, M.; Jadhav, S.M.; Majumdar, T.D.; Shete-Aich, A.; Basu, A.; Abraham, P.; et al. Full-Genome Sequences of the First Two SARS-CoV-2 Viruses from India. Indian J. Med. Res. 2020, 151, 200. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-NCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 10224. [Google Scholar] [CrossRef]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and Genomics of SARS-CoV-2: A Review of the Literature with the Special Focus on Genetic Diversity and SARS-CoV-2 Genome Detection. Genomics 2020, 113. [Google Scholar] [CrossRef]
- Roy, C.; Mandal, S.M.; Mondol, S.K.; Mukherjee, S.; Ghosh, W.; Chakraborty, R. Trends of Mutation Accumulation across Global SARS-CoV-2 Genomes: Implications for the Ecology and Evolution of the Novel Coronavirus. Genomics 2020. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Yadav, P.D.; Albariño, C.G.; Nyayanit, D.A.; Guerrero, L.; Jenks, M.H.; Sarkale, P.; Nichol, S.T.; Mourya, D.T. Equine Encephalosis Virus in India, 2008. Emerg. Infect. Dis. 2018, 24, 898–901. [Google Scholar] [CrossRef]
- Yadav, P.D.; Nyayanit, D.A.; Shete, A.M.; Jain, S.; Majumdar, T.P.; Chaubal, G.Y.; Shil, P.; Kore, P.M.; Mourya, D.T. Complete Genome Sequencing of Kaisodi Virus Isolated from Ticks in India Belonging to Phlebovirus Genus, Family Phenuiviridae. Ticks Tick-Borne Dis. 2019, 10, 23–33. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—from Vision to Reality. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Banu, S.; Jolly, B.; Mukherjee, P.; Singh, P.; Khan, S.; Zaveri, L.; Shambhavi, S.; Gaur, N.; Reddy, S.; Kaveri, K.; et al. A Distinct Phylogenetic Cluster of Indian SARS-CoV-2 Isolates. Open Forum Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T.; et al. Evolutionary and Structural Analyses of SARS-CoV-2 D614G Spike Protein Mutation Now Documented Worldwide. Sci. Rep. 2020, 10, 14031. [Google Scholar] [CrossRef]
- Potdar, V.; Cherian, S.S.; Deshpande, G.R.; Ullas, P.T.; Yadav, P.D.; Choudhary, M.L.; Gughe, R.; Vipat, V.; Jadhav, S.; Patil, S.; et al. Genomic Analysis of SARS-CoV-2 Strains among Indians Returning from Italy, Iran & China, & Italian Tourists in India. Indian J. Med. Res. 2020, 151, 255. [Google Scholar] [CrossRef]
- Coronavirus Disease (COVID-19) Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ (accessed on 22 August 2020).
- Leitmeyer, K.; Rico-Hesse, R. Viral Evolution and Epidemiology. Curr. Opin. Infect. Dis. 1997, 10, 367–371. [Google Scholar] [CrossRef]
- Parvez, M.K.; Parveen, S. Evolution and Emergence of Pathogenic Viruses: Past, Present, and Future. Intervirology 2017, 60, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Strang, C.; Ferguson, N.; Cori, A.; Jombart, T. When Are Pathogen Genome Sequences Informative of Transmission Events? PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef]
- GISAID—HCoV-19 Genomic Epidemiology. Available online: https://www.gisaid.org/epiflu-applications/hcov-19-genomic-epidemiology/ (accessed on 22 September 2020).
- Yadav, P.D.; Nyayanit, D.A.; Sahay, R.R.; Sarkale, P.; Pethani, J.; Patil, S.; Baradkar, S.; Potdar, V.; Patil, D.Y. Isolation and characterization of VUI-202012/01, a SARS-CoV-2 variant: Human cases travelled from United Kingdom to India. J. Travel Med. 2021, 28, taab009. [Google Scholar]
- Yadav, P.D.; Nyayanit, D.A.; Sahay, R.R.; Shete, A.M.; Majumdar, T.; Patil, S.; Patil, D.Y.; Gupta, N.; Kaur, H.; Aggarwal, N.; et al. Imported SARS-CoV-2 V501Y.V2 variant (B.1.351) detected in travelers from South Africa and Tanzania to India. Travel Med. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a Rapidly Evolving Immune Evasion Protein. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GISAID Clades | Percentage Evolutionary Divergence over Sequence Pairs between Groups | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| L | S | V | Unclassified Cluster | G | GR | GH | GH-GR | GV-GR | GS | |
| L | 0.081 | 0.077 | 0.070 | 0.077 | 0.108 | 0.101 | 0.112 | 0.152 | 0.097 | |
| S | 0.041 | 0.093 | 0.088 | 0.094 | 0.125 | 0.117 | 0.129 | 0.170 | 0.114 | |
| V | 0.043 | 0.050 | 0.100 | 0.090 | 0.122 | 0.114 | 0.125 | 0.166 | 0.074 | |
| Unclassified Cluster | 0.035 | 0.045 | 0.052 | 0.097 | 0.128 | 0.120 | 0.131 | 0.173 | 0.118 | |
| G | 0.043 | 0.051 | 0.053 | 0.051 | 0.080 | 0.072 | 0.085 | 0.124 | 0.084 | |
| GR | 0.058 | 0.066 | 0.068 | 0.066 | 0.047 | 0.104 | 0.069 | 0.100 | 0.115 | |
| GH | 0.059 | 0.067 | 0.069 | 0.066 | 0.047 | 0.063 | 0.087 | 0.149 | 0.105 | |
| GH-GR | 0.060 | 0.068 | 0.070 | 0.068 | 0.050 | 0.041 | 0.055 | 0.109 | 0.114 | |
| GV-GR | 0.077 | 0.085 | 0.087 | 0.085 | 0.066 | 0.052 | 0.083 | 0.056 | 0.160 | |
| GS | 0.054 | 0.062 | 0.040 | 0.063 | 0.050 | 0.065 | 0.061 | 0.064 | 0.085 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, P.D.; Nyayanit, D.A.; Majumdar, T.; Patil, S.; Kaur, H.; Gupta, N.; Shete, A.M.; Pandit, P.; Kumar, A.; Aggarwal, N.; et al. An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India. Viruses 2021, 13, 925. https://doi.org/10.3390/v13050925
Yadav PD, Nyayanit DA, Majumdar T, Patil S, Kaur H, Gupta N, Shete AM, Pandit P, Kumar A, Aggarwal N, et al. An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India. Viruses. 2021; 13(5):925. https://doi.org/10.3390/v13050925
Chicago/Turabian StyleYadav, Pragya D., Dimpal A. Nyayanit, Triparna Majumdar, Savita Patil, Harmanmeet Kaur, Nivedita Gupta, Anita M. Shete, Priyanka Pandit, Abhinendra Kumar, Neeraj Aggarwal, and et al. 2021. "An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India" Viruses 13, no. 5: 925. https://doi.org/10.3390/v13050925
APA StyleYadav, P. D., Nyayanit, D. A., Majumdar, T., Patil, S., Kaur, H., Gupta, N., Shete, A. M., Pandit, P., Kumar, A., Aggarwal, N., Narayan, J., Vijay, N., Kalawat, U., Sugunan, A. P., Munivenkatappa, A., Sharma, T., Devi, S., Majumdar, T., Jaryal, S., ... Bhargava, B. (2021). An Epidemiological Analysis of SARS-CoV-2 Genomic Sequences from Different Regions of India. Viruses, 13(5), 925. https://doi.org/10.3390/v13050925