
Visualizing Rous Sarcoma Virus Genomic RNA Dimerization in the Nucleus, Cytoplasm, and at the Plasma Membrane


Abstract
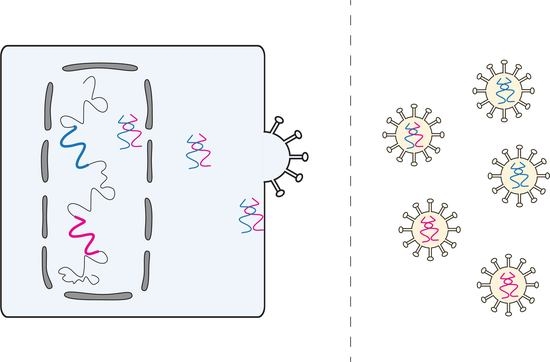
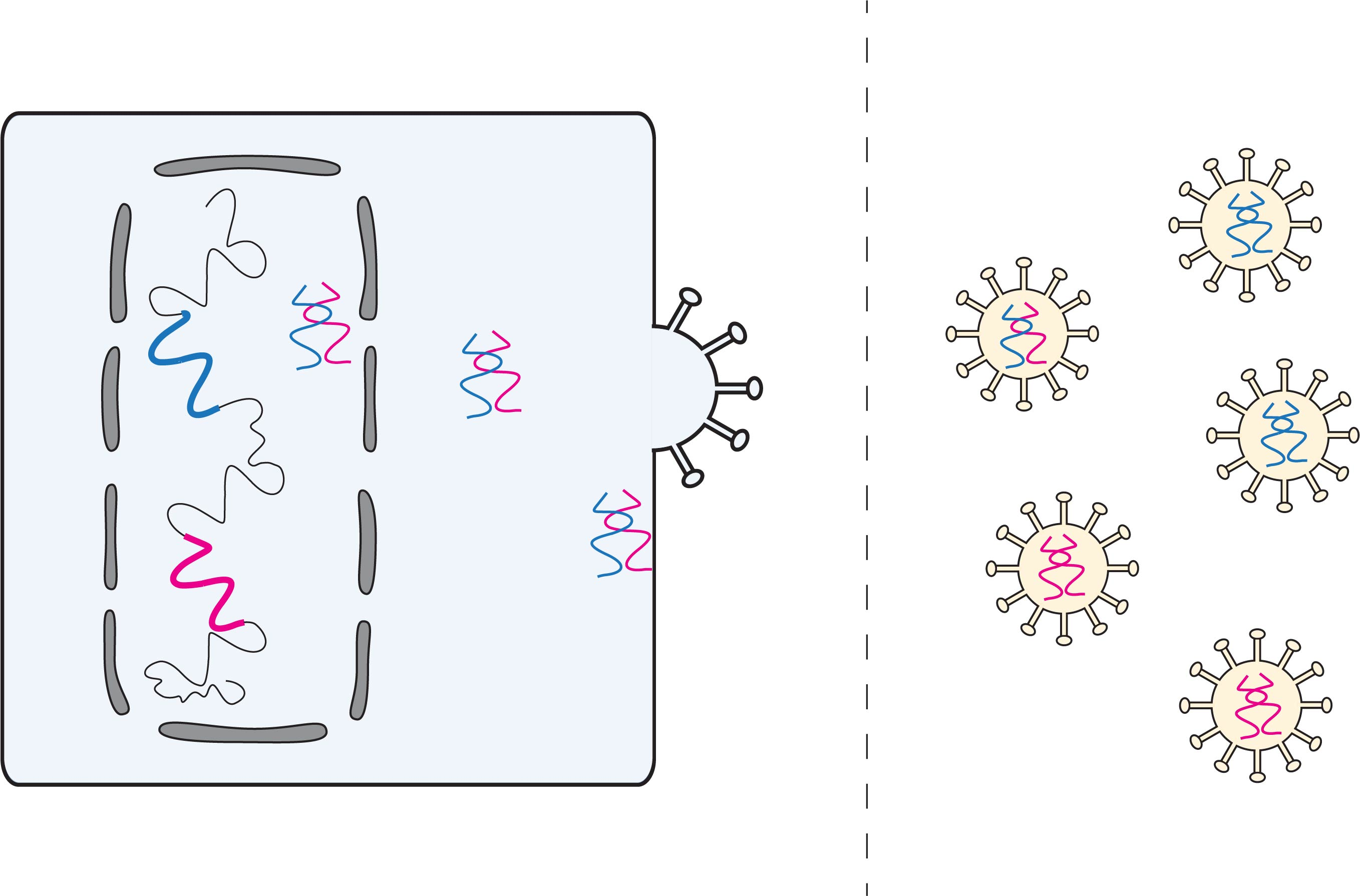
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Cloning
2.2. Cell Culture, Transfection, Slide Preparation, and Virus Particle Purification
2.3. Immunoblot Analysis
2.4. Probe Preparation and Single Molecule Fluorescence in Situ Hybridization (smFISH)
2.5. Microscopy, Image Acquisition, Image Analysis
3. Results
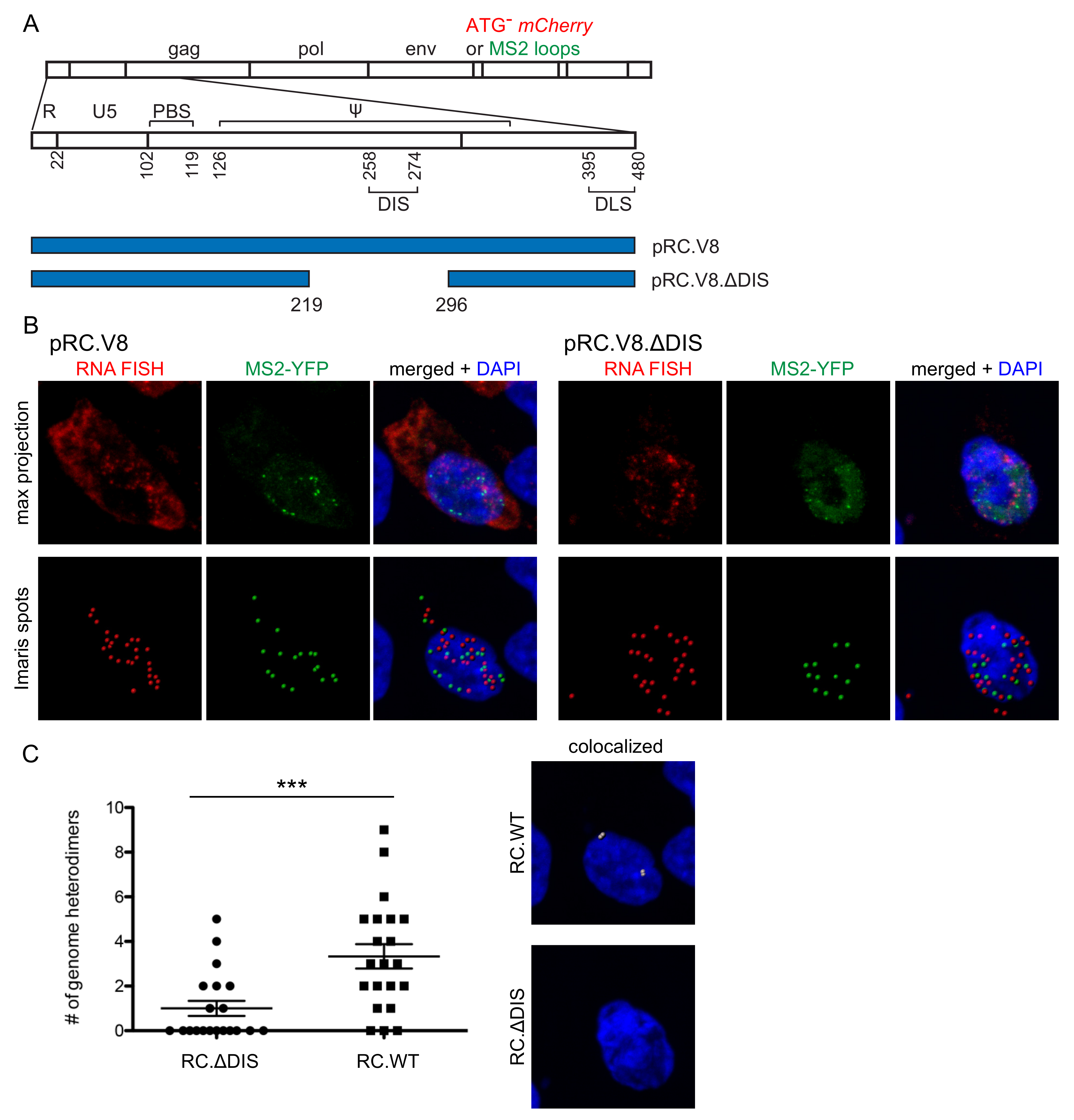
3.1. Three Imaging Combinations to Visualize Genome Heterodimers within Cells
3.2. Visualization of RSV Genome Heterodimers in Different Subcellular Compartments
3.3. Genomic RNA Heterodimer Formation Depends on the Presence of the DIS
3.4. Genome Dimer Preferences in RSV
3.5. Spliced vRNA Does Not Contribute to Intracellular Heterodimers in RSV
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berkhout, B.; van Wamel, J.L. Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 1996, 70, 6723–6732. [Google Scholar] [CrossRef] [PubMed]
- Garbitt, R.A.; Albert, J.A.; Kessler, M.D.; Parent, L.J. Trans-acting inhibition of genomic RNA dimerization by Rous sarcoma virus matrix mutants. J. Virol. 2001, 75, 260–268. [Google Scholar] [CrossRef]
- Laughrea, M.; Jette, L.; Mak, J.; Kleiman, L.; Liang, C.; Wainberg, M.A. Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol. 1997, 71, 3397–3406. [Google Scholar] [CrossRef]
- Parent, L.J.; Cairns, T.M.; Albert, J.A.; Wilson, C.B.; Wills, J.W.; Craven, R.C. RNA dimerization defect in a Rous sarcoma virus matrix mutant. J. Virol. 2000, 74, 164–172. [Google Scholar] [CrossRef]
- Sakuragi, J.; Ueda, S.; Iwamoto, A.; Shioda, T. Possible role of dimerization in human immunodeficiency virus type 1 genome RNA packaging. J. Virol. 2003, 77, 4060–4069. [Google Scholar] [CrossRef][Green Version]
- Houzet, L.; Paillart, J.C.; Smagulova, F.; Maurel, S.; Morichaud, Z.; Marquet, R.; Mougel, M. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 2007, 35, 2695–2704. [Google Scholar] [CrossRef]
- Dubois, N.; Marquet, R.; Paillart, J.C.; Bernacchi, S. Retroviral RNA Dimerization: From Structure to Functions. Front. Microbiol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Johnson, S.F.; Telesnitsky, A. Retroviral RNA dimerization and packaging: The what, how, when, where, and why. PLoS Pathog. 2010, 6, e1001007. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Hu, W.S. HIV-1 RNA dimerization: It takes two to tango. AIDS Rev. 2009, 11, 91–102. [Google Scholar] [PubMed]
- Paillart, J.C.; Shehu-Xhilaga, M.; Marquet, R.; Mak, J. Dimerization of retroviral RNA genomes: An inseparable pair. Nat. Rev. Microbiol. 2004, 2, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Temin, H.M. Genetic consequences of packaging two RNA genomes in one retroviral particle: Pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 1990, 87, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, J.G.; Pedersen, F.S. Genetic reassortment and patch repair by recombination in retroviruses. J. Biomed. Sci. 2000, 7, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Nikolaitchik, O.A.; Chen, J.; Hammarskjold, M.L.; Rekosh, D.; Hu, W.S. Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS Pathog. 2009, 5, e1000627. [Google Scholar] [CrossRef] [PubMed]
- Bender, W.; Chien, Y.H.; Chattopadhyay, S.; Vogt, P.K.; Gardner, M.B.; Davidson, N. High-molecular-weight RNAs of AKR, NZB, and wild mouse viruses and avian reticuloendotheliosis virus all have similar dimer structures. J. Virol. 1978, 25, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.J.; Hu, S.; Bender, W.; Bailey, J.M.; Davidson, N.; Nicolson, M.O.; McAllister, R.M. RD-114, baboon, and woolly monkey viral RNA’s compared in size and structure. Cell 1976, 7, 609–620. [Google Scholar] [CrossRef]
- Murti, K.G.; Bondurant, M.; Tereba, A. Secondary structural features in the 70S RNAs of Moloney murine leukemia and Rous sarcoma viruses as observed by electron microscopy. J. Virol. 1981, 37, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.E.; Tizard, R.; Gilbert, W. Nucleotide sequence of Rous sarcoma virus. Cell 1983, 32, 853–869. [Google Scholar] [CrossRef]
- Fosse, P.; Motte, N.; Roumier, A.; Gabus, C.; Muriaux, D.; Darlix, J.L.; Paoletti, J. A short autocomplementary sequence plays an essential role in avian sarcoma-leukosis virus RNA dimerization. Biochemistry 1996, 35, 16601–16609. [Google Scholar] [CrossRef]
- Haddrick, M.; Lear, A.L.; Cann, A.J.; Heaphy, S. Evidence that a kissing loop structure facilitates genomic RNA dimerisation in HIV-1. J. Mol. Biol. 1996, 259, 58–68. [Google Scholar] [CrossRef]
- Fu, W.; Gorelick, R.J.; Rein, A. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 1994, 68, 5013–5018. [Google Scholar] [CrossRef]
- Fu, W.; Rein, A. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 1993, 67, 5443–5449. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.M.; de Rocquigny, H.; Roques, B.P.; Paoletti, J. A model of PSI dimerization: Destabilization of the C278-G303 stem-loop by the nucleocapsid protein (NCp10) of MoMuLV. Biochemistry 1996, 35, 8705–8714. [Google Scholar] [CrossRef] [PubMed]
- Jalalirad, M.; Laughrea, M. Formation of immature and mature genomic RNA dimers in wild-type and protease-inactive HIV-1: Differential roles of the Gag polyprotein, nucleocapsid proteins NCp15, NCp9, NCp7, and the dimerization initiation site. Virology 2010, 407, 225–236. [Google Scholar] [CrossRef][Green Version]
- Muriaux, D.; De Rocquigny, H.; Roques, B.P.; Paoletti, J. NCp7 activates HIV-1Lai RNA dimerization by converting a transient loop-loop complex into a stable dimer. J. Bio Chem. 1996, 271, 33686–33692. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.A.; An, W.; King, S.R.; Telesnitsky, A. Nonrandom dimerization of murine leukemia virus genomic RNAs. J. Virol. 2004, 78, 12129–12139. [Google Scholar] [CrossRef]
- Flynn, J.A.; Telesnitsky, A. Two distinct Moloney murine leukemia virus RNAs produced from a single locus dimerize at random. Virology 2006, 344, 391–400. [Google Scholar] [CrossRef]
- Maurel, S.; Houzet, L.; Garcia, E.L.; Telesnitsky, A.; Mougel, M. Characterization of a natural heterodimer between MLV genomic RNA and the SD’ retroelement generated by alternative splicing. RNA 2007, 13, 2266–2276. [Google Scholar] [CrossRef] [PubMed]
- Maurel, S.; Mougel, M. Murine leukemia virus RNA dimerization is coupled to transcription and splicing processes. Retrovirology 2010, 7, 64. [Google Scholar] [CrossRef]
- Kharytonchyk, S.A.; Kireyeva, A.I.; Osipovich, A.B.; Fomin, I.K. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology 2005, 2, 3. [Google Scholar] [CrossRef]
- Chen, J.; Nikolaitchik, O.; Singh, J.; Wright, A.; Bencsics, C.E.; Coffin, J.M.; Ni, N.; Lockett, S.; Pathak, V.K.; Hu, W.S. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 13535–13540. [Google Scholar] [CrossRef]
- Chen, J.; Rahman, S.A.; Nikolaitchik, O.A.; Grunwald, D.; Sardo, L.; Burdick, R.C.; Plisov, S.; Liang, E.; Tai, S.; Pathak, V.K.; et al. HIV-1 RNA genome dimerizes on the plasma membrane in the presence of Gag protein. Proc. Natl. Acad. Sci. USA 2016, 113, e201–e208. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Clerte, C.; Chamontin, C.; Basyuk, E.; Laine, S.; Hottin, J.; Bertrand, E.; Margeat, E.; Mougel, M. Imaging HIV-1 RNA dimerization in cells by multicolor super-resolution and fluctuation microscopies. Nucleic Acids Res. 2016, 44, 7922–7934. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Visualizing HIV-1 assembly. J. Mol. Biol. 2011, 410, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Miyake, A.; Nomaguchi, M.; Adachi, A. Structural dynamics of retroviral genome and the packaging. Front. Microbiol. 2011, 2, 264. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.S.; Liang, C.; Wainberg, M.A. Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably? Retrovirology 2004, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nikolaitchik, O.A.; Dilley, K.A.; Fu, W.; Gorelick, R.J.; Tai, S.H.; Soheilian, F.; Ptak, R.G.; Nagashima, K.; Pathak, V.K.; Hu, W.S. Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 2013, 9, e1003249. [Google Scholar] [CrossRef]
- Clever, J.L.; Miranda, D., Jr.; Parslow, T.G. RNA structure and packaging signals in the 5′ leader region of the human immunodeficiency virus type 1 genome. J. Virol. 2002, 76, 12381–12387. [Google Scholar] [CrossRef]
- Nikolaitchik, O.A.; Somoulay, X.; Rawson, J.M.; Yoo, J.A.; Pathak, V.K.; Hu, W.S. Unpaired Guanosines in the 5′ Untranslated Region of HIV-1 RNA Act Synergistically To Mediate Genome Packaging. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Helga-Maria, C.; Hammarskjöld, M.L.; Rekosh, D. An intact TAR element and cytoplasmic localization are necessary for efficient packaging of human immunodeficiency virus type 1 genomic RNA. J. Virol. 1999, 73, 4127–4135. [Google Scholar] [CrossRef]
- Kaye, J.F.; Richardson, J.H.; Lever, A.M. Cis-acting sequences involved in human immunodeficiency virus type 1 RNA packaging. J. Virol. 1995, 69, 6588–6592. [Google Scholar] [CrossRef]
- Banks, J.D.; Kealoha, B.O.; Linial, M.L. An Mpsi-containing heterologous RNA, but not env mRNA, is efficiently packaged into avian retroviral particles. J. Virol. 1999, 73, 8926–8933. [Google Scholar] [CrossRef]
- Craven, R.C.; Leure-duPree, A.E.; Weldon, R.A.; Wills, J.W. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J. Virol. 1995, 69, 4213–4227. [Google Scholar] [CrossRef]
- Dong, J.Y.; Dubay, J.W.; Perez, L.G.; Hunter, E. Mutations within the proteolytic cleavage site of the Rous sarcoma virus glycoprotein define a requirement for dibasic residues for intracellular cleavage. J. Virol. 1992, 66, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, C.J.; Hughes, S.H. Replication-competent retrovirus vectors for the transfer and expression of gene cassettes in avian cells. J. Virol. 1991, 65, 3728–3737. [Google Scholar] [CrossRef]
- Hughes, S.; Kosik, E. Mutagenesis of the region between env and src of the SR-A strain of Rous sarcoma virus for the purpose of constructing helper-independent vectors. Virology 1984, 136, 89–99. [Google Scholar] [CrossRef]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef]
- Larson, D.R.; Zenklusen, D.; Wu, B.; Chao, J.A.; Singer, R.H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 2011, 332, 475–478. [Google Scholar] [CrossRef]
- Fusco, D.; Accornero, N.; Lavoie, B.; Shenoy, S.M.; Blanchard, J.M.; Singer, R.H.; Bertrand, E. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 2003, 13, 161–167. [Google Scholar] [CrossRef]
- Chen, J.; Grunwald, D.; Sardo, L.; Galli, A.; Plisov, S.; Nikolaitchik, O.A.; Chen, D.; Lockett, S.; Larson, D.R.; Pathak, V.K.; et al. Cytoplasmic HIV-1 RNA is mainly transported by diffusion in the presence or absence of Gag protein. Proc. Natl. Acad. Sci. USA 2014, 111, e5205–e5213. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kaddis Maldonado, R.; Rye-McCurdy, T.; Binkley, C.; Bah, A.; Chen, E.C.; Rice, B.L.; Parent, L.J.; Musier-Forsyth, K. Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity. Viruses 2020, 12, 568. [Google Scholar] [CrossRef] [PubMed]
- Moscovici, C.; Moscovici, M.G.; Jimenez, H.; Lai, M.M.; Hayman, M.J.; Vogt, P.K. Continuous tissue culture cell lines derived from chemically induced tumors of Japanese quail. Cell 1977, 11, 95–103. [Google Scholar] [CrossRef]
- Matic, I.; van Hagen, M.; Schimmel, J.; Macek, B.; Ogg, S.C.; Tatham, M.H.; Hay, R.T.; Lamond, A.I.; Mann, M.; Vertegaal, A.C.O. In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell. Proteom. 2008, 7, 132–144. [Google Scholar] [CrossRef]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef]
- Hocine, S.; Raymond, P.; Zenklusen, D.; Chao, J.A.; Singer, R.H. Single-molecule analysis of gene expression using two-color RNA labeling in live yeast. Nat. Methods 2013, 10, 119–121. [Google Scholar] [CrossRef]
- Lim, F.; Peabody, D.S. RNA recognition site of PP7 coat protein. Nucleic Acids Res. 2002, 30, 4138–4144. [Google Scholar] [CrossRef]
- Polge, E.; Darlix, J.L.; Paoletti, J.; Fosse, P. Characterization of loose and tight dimer forms of avian leukosis virus RNA. J. Mol. Biol. 2000, 300, 41–56. [Google Scholar] [CrossRef]
- Lear, A.L.; Haddrick, M.; Heaphy, S. A study of the dimerization of Rous sarcoma virus RNA in vitro and in vivo. Virology 1995, 212, 47–57. [Google Scholar] [CrossRef]
- Darlix, J.L.; Zuker, M.; Spahr, P.F. Structure-function relationship of Rous sarcoma virus leader RNA. Nucleic Acids Res. 1982, 10, 5183–5196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bieth, E.; Gabus, C.; Darlix, J.L. A study of the dimer formation of Rous sarcoma virus RNA and of its effect on viral protein synthesis in vitro. Nucleic Acids Res. 1990, 18, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Rabson, A.B.; Graves, B.J. Processing of Retroviral RNA. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Garbitt, R.A.; Bone, K.R.; Parent, L.J. Insertion of a classical nuclear import signal into the matrix domain of the Rous sarcoma virus Gag protein interferes with virus replication. J. Virol. 2004, 78, 13534–13542. [Google Scholar] [CrossRef]
- Garbitt-Hirst, R.; Kenney, S.P.; Parent, L.J. Genetic evidence for a connection between Rous sarcoma virus gag nuclear trafficking and genomic RNA packaging. J. Virol. 2009, 83, 6790–6797. [Google Scholar] [CrossRef]
- Gudleski, N.; Flanagan, J.M.; Ryan, E.P.; Bewley, M.C.; Parent, L.J. Directionality of nucleocytoplasmic transport of the retroviral gag protein depends on sequential binding of karyopherins and viral RNA. Proc. Natl. Acad. Sci. USA 2010, 107, 9358–9363. [Google Scholar] [CrossRef]
- Scheifele, L.Z.; Garbitt, R.A.; Rhoads, J.D.; Parent, L.J. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc. Natl. Acad. Sci. USA 2002, 99, 3944–3949. [Google Scholar] [CrossRef]
- Scheifele, L.Z.; Kenney, S.P.; Cairns, T.M.; Craven, R.C.; Parent, L.J. Overlapping roles of the Rous sarcoma virus Gag p10 domain in nuclear export and virion core morphology. J. Virol. 2007, 81, 10718–10728. [Google Scholar] [CrossRef]
- Scheifele, L.Z.; Ryan, E.P.; Parent, L.J. Detailed mapping of the nuclear export signal in the Rous sarcoma virus Gag protein. J. Virol. 2005, 79, 8732–8741. [Google Scholar] [CrossRef]
- Maldonado, R.J.K.; Rice, B.; Chen, E.C.; Tuffy, K.M.; Chiari, E.F.; Fahrbach, K.M.; Hope, T.J.; Parent, L.J. Visualizing Association of the Retroviral Gag Protein with Unspliced Viral RNA in the Nucleus. mBio 2020, 11. [Google Scholar] [CrossRef]
- Blair, D. Genetic recombination between avian leukosis and sarcoma viruses Experimental variables and the frequencies of recombination. Virology 1977, 77, 534–544. [Google Scholar] [CrossRef]
- Ortiz-Conde, B.A.; Hughes, S.H. Studies of the genomic RNA of leukosis viruses: Implications for RNA dimerization. J. Virol. 1999, 73, 7165–7174. [Google Scholar] [CrossRef] [PubMed]
- Meric, C.; Gouilloud, E.; Spahr, P.F. Mutations in Rous sarcoma virus nucleocapsid protein p12 (NC): Deletions of Cys-His boxes. J. Virol. 1988, 62, 3328–3333. [Google Scholar] [CrossRef] [PubMed]
- Meric, C.; Spahr, P.F. Rous sarcoma virus nucleic acid-binding protein p12 is necessary for viral 70S RNA dimer formation and packaging. J. Virol. 1986, 60, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.M.; Vogt, V.M. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J. Virol. 2004, 78, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Cawte, A.D.; Unrau, P.J.; Rueda, D.S. Live cell imaging of single RNA molecules with fluorogenic Mango II arrays. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Lu, K.; Heng, X.; Garyu, L.; Monti, S.; Garcia, E.L.; Kharytonchyk, S.; Dorjsuren, B.; Kulandaivel, G.; Jones, S.; Hiremath, A.; et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 2011, 334, 242–245. [Google Scholar] [CrossRef]
- Tuffy, K.M.; Maldonado, R.J.K.; Chang, J.; Rosenfeld, P.; Cochrane, A.; Parent, L.J. HIV-1 gag forms ribonucleoprotein complexes with unspliced viral RNA at transcription sites. Viruses 2020, 12, 1281. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mCherry smFISH + PP7 smFISH | mCherry smFISH + MS2-YFP | MS2-mCherry + Bgl-YFP | |
|---|---|---|---|
| Total red non-colocalized foci | 590 | 1063 | 504 |
| Total green non-colocalized foci | 792 | 446 | 940 |
| Total red/green colocalized foci | 92 | 153 | 283 |
| Total number of foci analyzed | 1474 | 1662 | 1727 |
| Total % red non-colocalized foci | 40.0% | 64.0% | 29.2% |
| Total % green non-colocalized foci | 53.7% | 26.8% | 54.4% |
| Total % red/green colocalized foci | 6.2% | 9.2% | 16.4% |
| Total % colocalized in nucleus | 67.4% | 50.3% | 47.0% |
| Total % colocalized in cytoplasm | 28.3% | 45.8% | 51.2% |
| Total % colocalized at plasma membrane | 4.3% | 3.9% | 1.8% |
| # cells | 24 | 27 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, E.C.; Maldonado, R.J.K.; Parent, L.J. Visualizing Rous Sarcoma Virus Genomic RNA Dimerization in the Nucleus, Cytoplasm, and at the Plasma Membrane. Viruses 2021, 13, 903. https://doi.org/10.3390/v13050903
Chen EC, Maldonado RJK, Parent LJ. Visualizing Rous Sarcoma Virus Genomic RNA Dimerization in the Nucleus, Cytoplasm, and at the Plasma Membrane. Viruses. 2021; 13(5):903. https://doi.org/10.3390/v13050903
Chicago/Turabian StyleChen, Eunice C., Rebecca J. Kaddis Maldonado, and Leslie J. Parent. 2021. "Visualizing Rous Sarcoma Virus Genomic RNA Dimerization in the Nucleus, Cytoplasm, and at the Plasma Membrane" Viruses 13, no. 5: 903. https://doi.org/10.3390/v13050903
APA StyleChen, E. C., Maldonado, R. J. K., & Parent, L. J. (2021). Visualizing Rous Sarcoma Virus Genomic RNA Dimerization in the Nucleus, Cytoplasm, and at the Plasma Membrane. Viruses, 13(5), 903. https://doi.org/10.3390/v13050903