
The Role of Non-Structural Protein NSs in the Pathogenesis of Severe Fever with Thrombocytopenia Syndrome


Abstract
1. Introduction
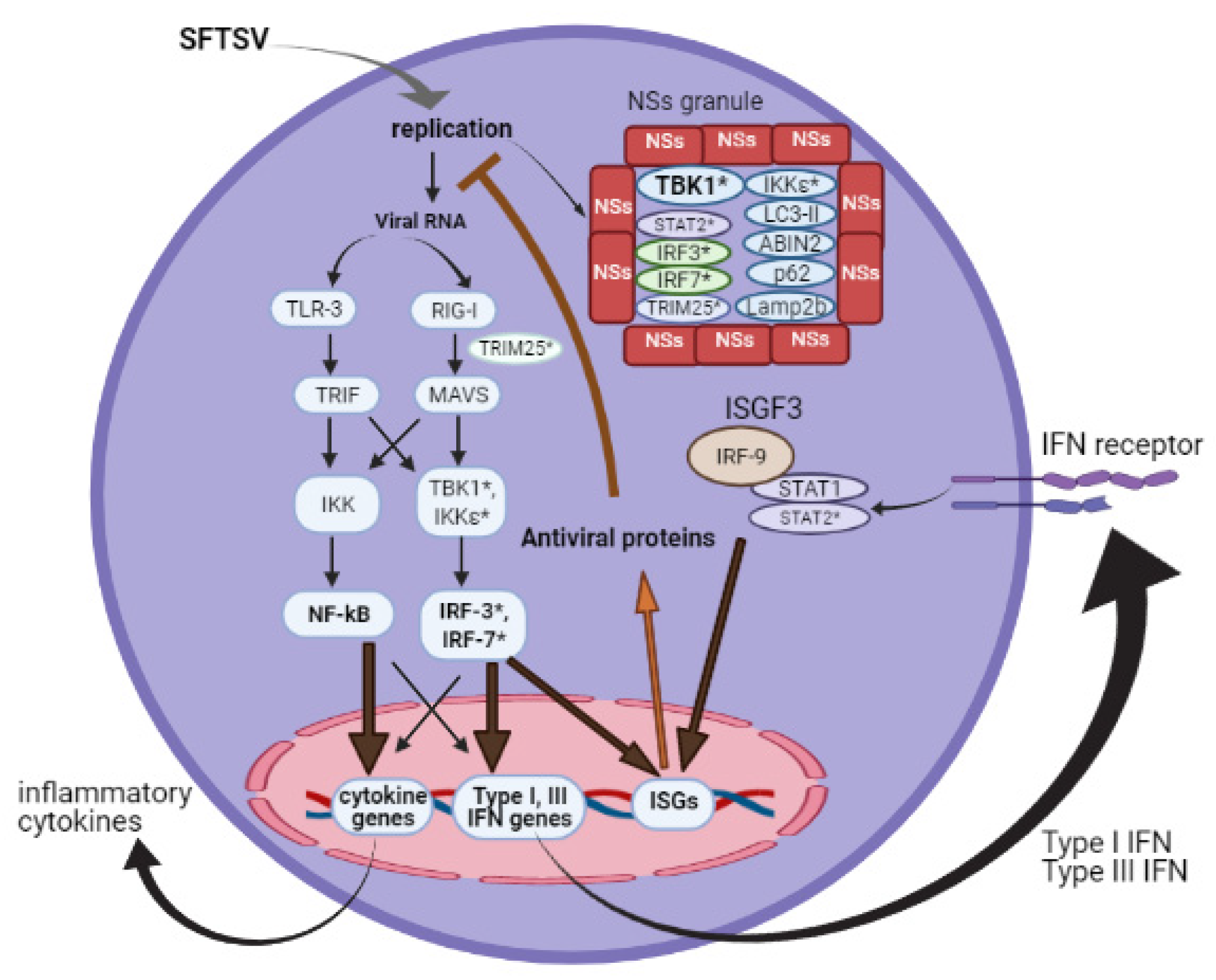
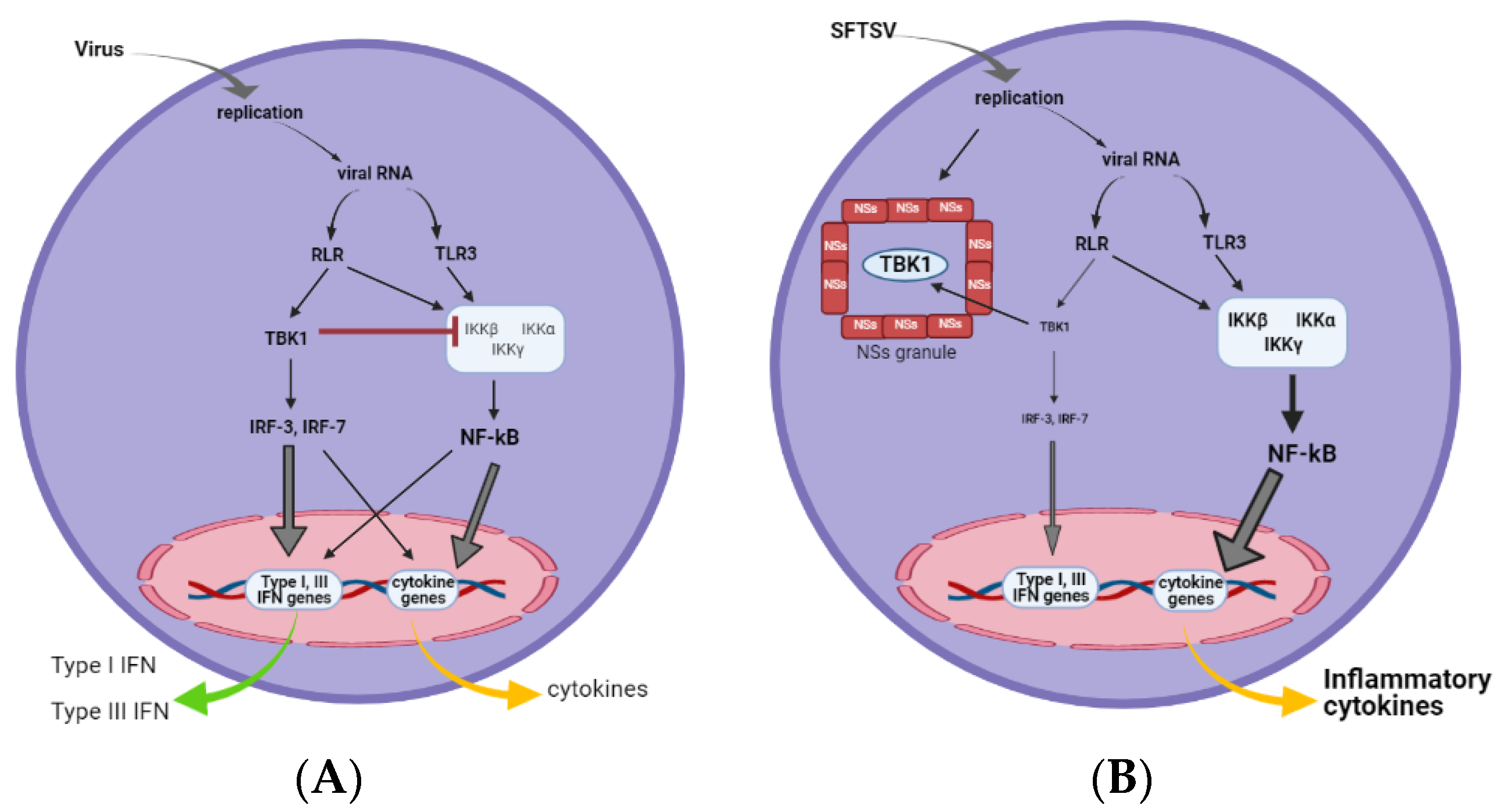
2. Influence of NSs on Innate Immunity
2.1. Induction of Interferon
2.2. Action of Interferon
2.3. Non-IFN Antiviral Responses
2.4. Role of NSs in Viral Replication
3. SFTSV Infection Mouse Models
4. Cytokine Storm Induction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunya-virales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Wang, Q.; Cheng, J.; Hu, B.; Li, J.; Zhan, F.; Song, Y.; Guo, D. Current status of severe fever with thrombocytopenia syndrome in China. Virol. Sin. 2017, 32, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Yamagishi, T.; Shimada, T.; Matsui, T.; Shimojima, M.; Saijo, M.; Oishi, K. Epidemiological and Clinical Features of Severe Fever with Thrombocytopenia Syndrome in Japan, 2013–2014. PLoS ONE 2016, 11, e0165207. [Google Scholar] [CrossRef] [PubMed]
- Word Health Organization (WHO). Annual Review of Diseases Prioritized under the Research and Development Blueprint. 2017. Available online: http://www.who.int/csr/research-and-development/en/ (accessed on 2 July 2019).
- Saijo, M. Pathophysiology of severe fever with thrombocytopenia syndrome and development of specific antiviral therapy. J. Infect. Chemother. 2018, 24, 773–781. [Google Scholar] [CrossRef]
- Reece, L.M.; Beasley, D.W.; Milligan, G.N.; Sarathy, V.V.; Barrett, A.D. Current status of Severe Fever with Thrombocytopenia Syndrome vaccine development. Curr. Opin. Virol. 2018, 29, 72–78. [Google Scholar] [CrossRef]
- Lin, F.-C.; Young, H.A. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Boisson-Dupuis, S.; Chapgier, A.; Yang, K.; Bustamante, J.; Puel, A.; Picard, C.; Abel, L.; Jouanguy, E.; Casanova, J.L. Inborn errors of interferon (IFN)-mediated immunity in humans: Insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol. Rev. 2008, 226, 29–40. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Kato, H.; Fujita, T. Sensing viral invasion by RIG-I like receptors. Curr. Opin. Microbiol. 2014, 20, 131–138. [Google Scholar] [CrossRef]
- Yamada, S.; Shimojima, M.; Narita, R.; Tsukamoto, Y.; Kato, H.; Saijo, M.; Fujita, T. RIG-I-Like Receptor and Toll-Like Receptor Sig-naling Pathways Cause Aberrant Production of Inflammatory Cytokines/Chemokines in a Severe Fever with Thrombo-cytopenia Syndrome Virus Infection Mouse Model. J. Virol. 2018, 92, e02246-17. [Google Scholar] [CrossRef]
- Min, Y.-Q.; Ning, Y.-J.; Wang, H.; Deng, F. A RIG-I–like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J. Biol. Chem. 2020, 295, 9691–9711. [Google Scholar] [CrossRef]
- Hong, Y.; Bai, M.; Qi, X.; Li, C.; Liang, M.; Li, D.; Cardona, C.J.; Xing, Z. Suppression of the IFN-α and -β Induction through Sequestering IRF7 into Viral Inclusion Bodies by Nonstructural Protein NSs in Severe Fever with Thrombocytopenia Syndrome Bunyavirus Infection. J. Immunol. 2019, 202, 841–856. [Google Scholar] [CrossRef]
- Qu, B.; Qi, X.; Wu, X.; Liang, M.; Li, C.; Cardona, C.J.; Xu, W.; Tang, F.; Li, Z.; Wu, B.; et al. Suppression of the interferon and NF-κB responses by severe fever with thrombocytopenia syndrome virus. J. Virol. 2012, 86, 8388–8401. [Google Scholar] [CrossRef]
- Wu, X.; Qi, X.; Qu, B.; Zhang, Z.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Evasion of antiviral immunity through sequestering of TBK1/IKKε/IRF3 into viral inclusion bodies. J. Virol. 2014, 88, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, R.; Sakabe, S.; Urata, S.; Yasuda, J. Species-Specific Pathogenicity of Severe Fever with Thrombocytopenia Syn-drome Virus Is Determined by Anti-STAT2 Activity of NSs. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Sakai, M.; Shimojima, M.; Saijo, M.; Itoh, M.; Gotoh, B. Nonstructural protein of severe fever with thrombocyto-penia syndrome phlebovirus targets STAT2 and not STAT1 to inhibit type I interferon-stimulated JAK-STAT signaling. Microbes Infect. 2018, 20, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.-J.; Feng, K.; Min, Y.-Q.; Cao, W.-C.; Wang, M.; Deng, F.; Hu, Z.; Wang, H. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J. Virol. 2015, 89, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.-J.; Mo, Q.; Feng, K.; Min, Y.-Q.; Li, M.; Hou, D.; Peng, C.; Zheng, X.; Deng, F.; Hu, Z.; et al. Interferon-γ-Directed Inhibition of a Novel High-Pathogenic Phlebovirus and Viral Antagonism of the Antiviral Signaling by Targeting STAT1. Front. Immunol. 2019, 10, 1182. [Google Scholar] [CrossRef]
- Rezelj, V.V.; Li, P.; Chaudhary, V.; Elliott, R.M.; Jin, D.Y.; Brennan, B. Differential Antagonism of Human Innate Immune Re-sponses by Tick-Borne Phlebovirus Nonstructural Proteins. mSphere 2017, 2, e00234-17. [Google Scholar] [CrossRef]
- Deng, B.; Zhang, S.; Geng, Y.; Zhang, Y.; Wang, Y.; Yao, W.; Wen, Y.; Cui, W.; Zhou, Y.; Gu, Q.; et al. Cytokine and Chemokine Levels in Patients with Severe Fever with Thrombocytopenia Syndrome Virus. PLoS ONE 2012, 7, e41365. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.-S.; Kim, M.-C.; Kim, J.Y.; Jeon, N.-Y.; Ryu, B.-H.; Hong, J.; Chong, Y.P.; Lee, S.-O.; Choi, S.-H.; Kim, Y.S.; et al. Kinetics of viral load and cytokines in severe fever with thrombocytopenia syndrome. J. Clin. Virol. 2018, 101, 57–62. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, M.-M.; Lei, X.-Y.; Yu, X.-J. SFTS phlebovirus promotes LC3-II accumulation and nonstructural protein of SFTS phlebovirus co-localizes with autophagy proteins. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Jiang, Z.; Shin, W.-J.; Jung, J.U. Severe Fever with Thrombocytopenia Syndrome Virus NSs Interacts with TRIM21 To Activate the p62-Keap1-Nrf2 Pathway. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated anti-microbial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Sun, Q.; Qi, X.; Zhang, Y.; Wu, X.; Liang, M.; Li, C.; Li, D.; Cardona, C.J.; Xing, Z. Synaptogyrin-2 Promotes Replication of a Novel Tick-borne Bunya-virus through Interacting with Viral Nonstructural Protein NSs. J. Biol. Chem. 2016, 291, 16138–16149. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, X.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. FASEB J. 2014, 28, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Kang, J.; Xu, L.; Zhang, K.; Li, X.; Hou, W.; Wang, Z.; Wang, T. The Severe Fever with Thrombocytopenia Syndrome Virus NSs Protein Interacts with CDK1 To Induce G2 Cell Cycle Arrest and Positively Regulate Viral Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, B.; Paessler, S.; Walker, D.H.; Tesh, R.B.; Yu, X.J. The pathogenesis of severe fever with thrombocytopenia syndrome virus infection in alpha/beta interferon knockout mice: Insights into the pathologic mechanisms of a new viral hemor-rhagic fever. J. Virol. 2014, 88, 1781–1786. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, C.; Zhan, F.; Wang, X.; Liang, M.; Zhang, Q.; Ding, S.; Guan, X.; Huo, X.; Li, C.; et al. Host Cytokine Storm Is Associated With Disease Severity of Severe Fever With Thrombocytopenia Syndrome. J. Infect. Dis. 2012, 206, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-P.; Liang, M.-F.; Ye, J.-B.; Liu, Q.-H.; Xiong, C.-H.; Long, B.; Lin, W.-B.; Cui, N.; Zou, Z.-Q.; Song, Y.-L.; et al. Prognostic value of clinical and immunological markers in acute phase of SFTS virus infection. Clin. Microbiol. Infect. 2014, 20, O870–O878. [Google Scholar] [CrossRef]
- Khalil, J.; Yamada, S.; Tsukamoto, Y.; Abe, H.; Shimojima, M.; Kato, H.; Fujita, T. The Non-structural Protein NSs of SFTSV Causes Cytokine Storm Through the Hyper-activation of NF-κB. Mol. Cell. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Park, S.-J.; Sun, Y.; Yoo, J.-S.; Pudupakam, R.S.; Foo, S.-S.; Shin, W.-J.; Chen, S.B.; Tsichlis, P.N.; Lee, W.-J.; et al. Severe fever with thrombocytopenia syndrome phlebovirus non-structural protein activates TPL2 signalling pathway for viral immunopathogenesis. Nat. Microbiol. 2019, 4, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zheng, N.; Liu, Y.; Tian, C.; Wu, X.; Ma, X.; Chen, D.; Zou, X.; Wang, G.; Wang, H.; et al. Deficient humoral responses and disrupted B-cell immunity are asso-ciated with fatal SFTSV infection. Nat. Commun. 2018, 9, 3328. [Google Scholar] [CrossRef]
- Zhang, L.; Fu, Y.; Wang, H.; Guan, Y.; Zhu, W.; Guo, M.; Zheng, N.; Wu, Z. Severe Fever With Thrombocytopenia Syndrome Virus-Induced Macrophage Differentiation Is Regulated by miR-146. Front. Immunol. 2019, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Léger, P.; Nachman, E.; Richter, K.; Tamietti, C.; Koch, J.; Burk, R.; Kummer, S.; Xin, Q.; Stanifer, M.; Bouloy, M.; et al. NSs amyloid formation is associated with the virulence of Rift Valley fever virus in mice. Nat. Commun. 2020, 11, 3281. [Google Scholar] [CrossRef] [PubMed]
- Struthers, J.K.; Swanepoel, R. Identification of a Major Non-structural Protein in the Nuclei of Rift Valley Fever Virus-infected Cells. J. Gen. Virol. 1982, 60, 381–384. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Zhang, Y.-L.; Xin, Q.-L.; Guan, Z.-Q.; Chen, X.; Zhang, X.-A.; Li, X.-K.; Xiao, G.-F.; Lozach, P.-Y.; et al. SFTSV Infection Induces BAK/BAX-Dependent Mitochondrial DNA Release to Trigger NLRP3 Inflammasome Activation. Cell Rep. 2020, 30, 4370–4385.e7. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, F.; Wang, L.; Gao, M.; Xie, Y.; Sun, Y.; Liu, H.; Yuan, Y.; Yi, W.; Huang, Z.; et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics 2020, 10, 12223–12240. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.; Bibbs, L.; Ulevitch, R. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Chen, X.; Hao, A.; Li, X.; Ye, K.; Zhao, C.; Yang, H.; Ma, H.; Hu, L.; Zhao, Z.; Hu, L.; et al. Activation of JNK and p38 MAPK Mediated by ZDHHC17 Drives Glioblastoma Multiforme Development and Malignant Progression. Theranostics 2020, 10, 998–1015. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Effect of Interaction |
|---|---|
| TBK1 | Suppressed IFN responses and activated NF-κB signaling [14,32] |
| LC3-II | Promoted viral replication and autophagy regulation during SFTSV infection [23] |
| TRIM25 | Inhibited ubiquitination and activation of RIG-I [11] |
| IRF3 | Inhibited transcription of IFN-I [14] |
| IRF7 | Inhibited transcription of IFN-I [12] |
| STAT2 | Suppressed JAK/STAT signaling and abrogated ISG production [16,17] |
| IKKε | Suppressed IFN responses [14] |
| CDK1 | Cell cycle arrest at G2/M transition [28] |
| ABIN2 | Activated TPL2 signaling and IL-10 production [33] |
| p62 | Unknown role in autophagy regulation [24] |
| Lamp2b | Unknown role in autophagy regulation [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, J.; Kato, H.; Fujita, T. The Role of Non-Structural Protein NSs in the Pathogenesis of Severe Fever with Thrombocytopenia Syndrome. Viruses 2021, 13, 876. https://doi.org/10.3390/v13050876
Khalil J, Kato H, Fujita T. The Role of Non-Structural Protein NSs in the Pathogenesis of Severe Fever with Thrombocytopenia Syndrome. Viruses. 2021; 13(5):876. https://doi.org/10.3390/v13050876
Chicago/Turabian StyleKhalil, Jumana, Hiroki Kato, and Takashi Fujita. 2021. "The Role of Non-Structural Protein NSs in the Pathogenesis of Severe Fever with Thrombocytopenia Syndrome" Viruses 13, no. 5: 876. https://doi.org/10.3390/v13050876
APA StyleKhalil, J., Kato, H., & Fujita, T. (2021). The Role of Non-Structural Protein NSs in the Pathogenesis of Severe Fever with Thrombocytopenia Syndrome. Viruses, 13(5), 876. https://doi.org/10.3390/v13050876