
Role of Transportin-SR2 in HIV-1 Nuclear Import


 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
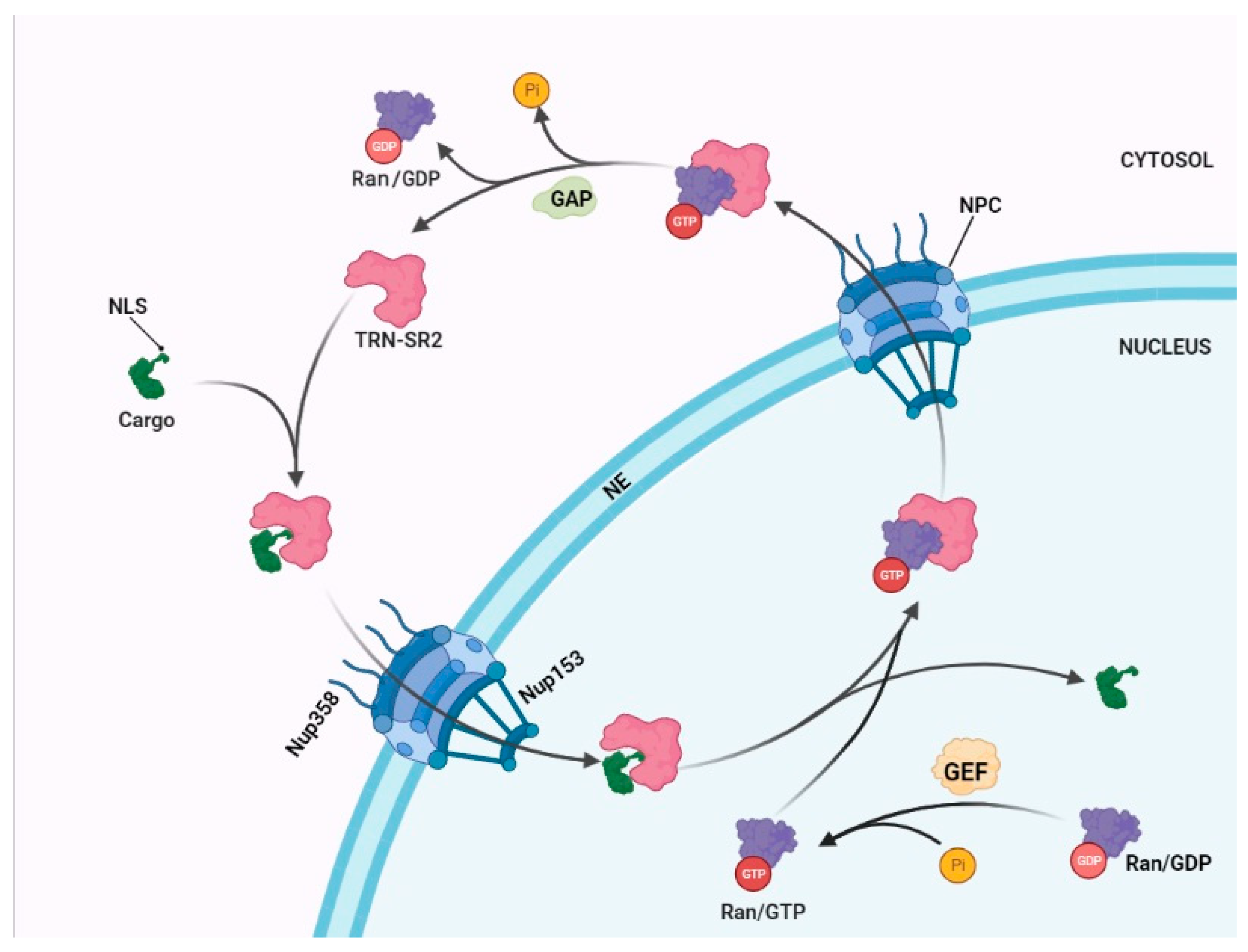
2. The Mechanism of a Nuclear Import
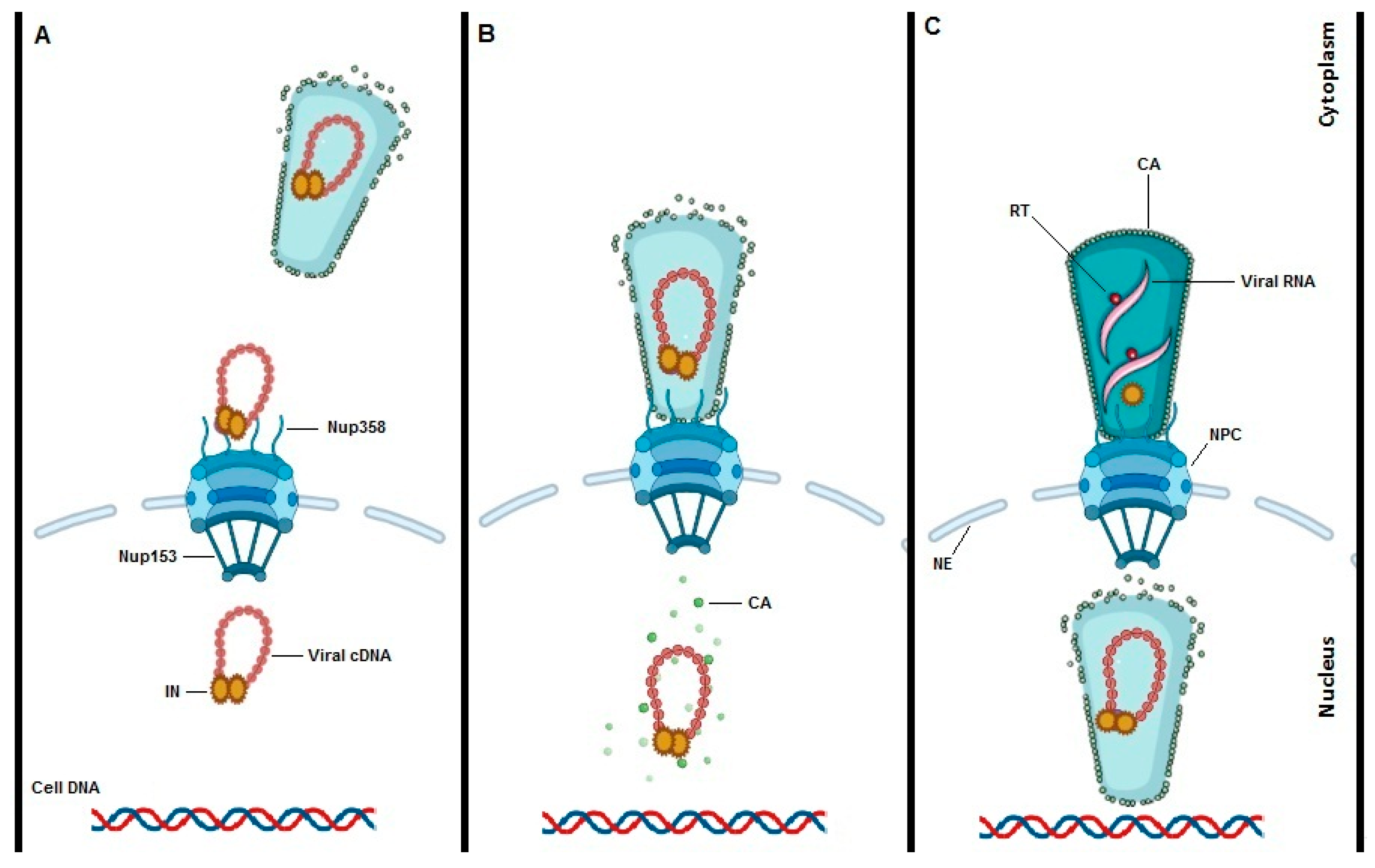
3. Transportin-SR2 Mediates Nuclear Import
4. Interplay between HIV-1 Integrase (IN) and TRN-SR2
5. Interaction between HIV-1 Capsid and TRN-SR2
6. Interplay between TRN-SR2 and CPSF6
7. Cyclophilin A and HIV-1 Nuclear Import
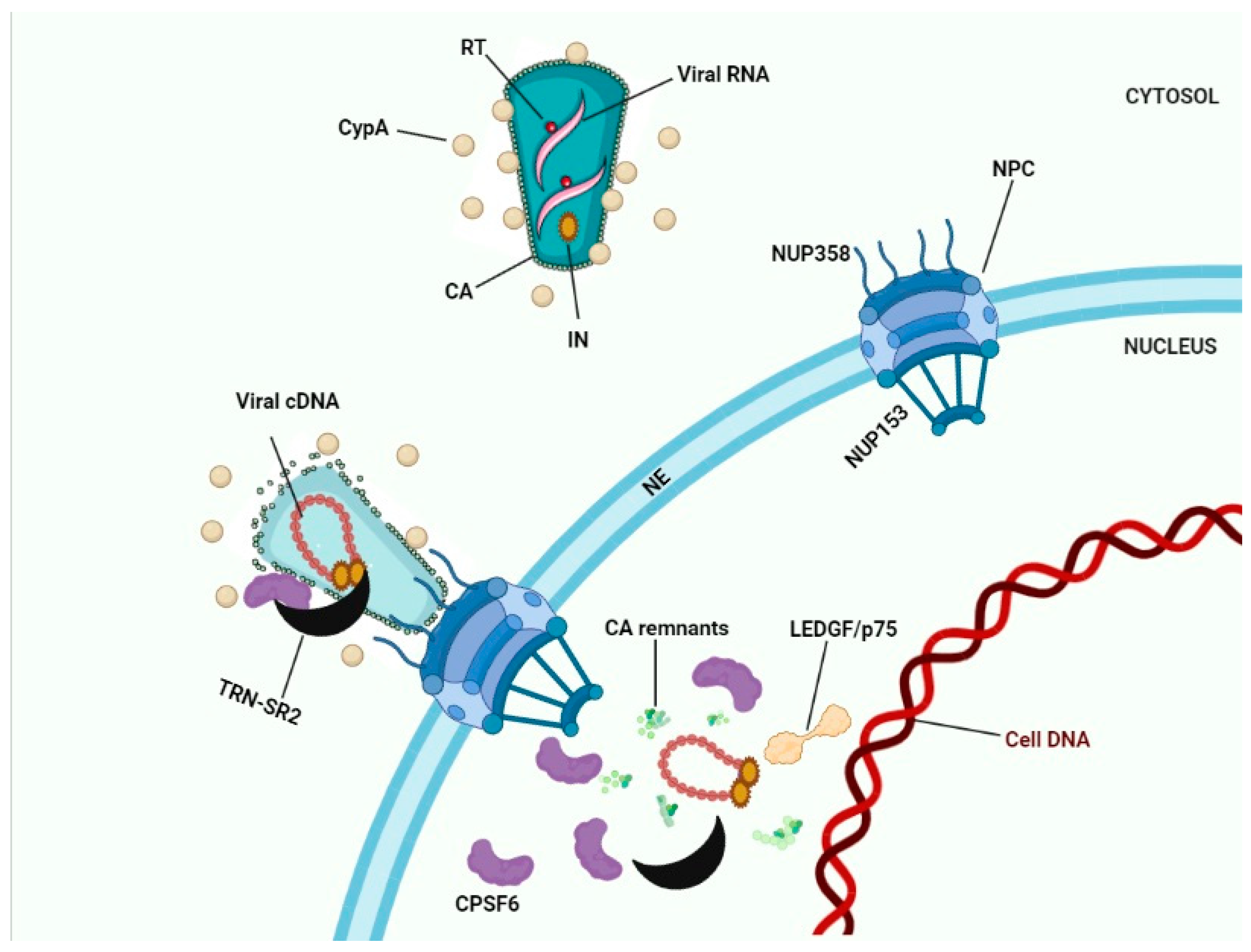
8. Conclusions: Towards a Model for HIV-1 Nuclear Entry
Funding
Conflicts of Interest
References
- Mettenleiter, T.C. Breaching the barrier—The nuclear envelope in virus infection. J. Mol. Biol. 2016, 428, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, L.; Kann, M. Nuclear import of hepatitis B virus capsids and genome. Viruses 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Greger, J.G.; Boimel, P.; Skalka, A.M. Human immunodeficiency virus type 1 DNA nuclear import and integration are mitosis independent in cycling cells. J. Virol. 2003, 77, 13412–13417. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harbor Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Keminer, O.; Peters, R. Permeability of single nuclear pores. Biophys. J. 1999, 77, 217–228. [Google Scholar] [CrossRef]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.-C.; Benarous, R.; Cereseto, A. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef]
- Krishnan, L.; Matreyek, K.A.; Oztop, I.; Lee, K.; Tipper, C.H.; Li, X.; Dar, M.J.; KewalRamani, V.N.; Engelman, A. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol. 2010, 84, 397–406. [Google Scholar] [CrossRef]
- Arnold, M.; Nath, A.; Hauber, J.; Kehlenbach, R.H. Multiple importins function as nuclear transport receptors for the Rev protein of human immunodeficiency virus type 1. J. Biol. Chem. 2006, 281, 20883–20890. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.; Swingler, S.; Aiken, C.; Trono, D. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 1995, 80, 379–388. [Google Scholar] [CrossRef][Green Version]
- Heinzinger, N.K.; Bukinsky, M.; Haggerty, S.A.; Ragland, A.M.; Kewalramani, V.; Lee, M.-A.; Gendelman, H.E.; Ratner, L.; Stevenson, M.; Emerman, M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7311–7315. [Google Scholar] [CrossRef] [PubMed]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef]
- Görlich, D.; Kutay, U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.; Bono, F.; Jinek, M.; Conti, E. Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 2007, 76, 647–671. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Danckaert, A.; Fricke, T.; Perez, P.; Fernandez, J.; Perret, E.; Roux, P.; Shorte, S.; Charneau, P.; Diaz-Griffero, F. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS ONE 2012, 7, e46037. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Yücel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Görlich, D.; Pante, N.; Kutay, U.; Aebi, U.; Bischoff, F. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996, 15, 5584–5594. [Google Scholar] [CrossRef]
- Ström, A.-C.; Weis, K. Importin-beta-like nuclear transport receptors. Genome Biol. 2001, 2, reviews3008. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, L.F.; Paschal, B.M. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005, 6, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, N.; Bachorik, J.L.; Dreyfuss, G. Transportin-SR, a nuclear import receptor for SR proteins. J. Cell Biol. 1999, 145, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N.; Cook, N.J.; Wang, W.; Hare, S.; Gupta, S.S.; Öztop, I.; Lee, K.; Pye, V.E.; Cosnefroy, O.; Snijders, A.P. Structural basis for nuclear import of splicing factors by human Transportin 3. Proc. Natl. Acad. Sci. USA 2014, 111, 2728–2733. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.-Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef]
- Ballandras-Colas, A.; Maskell, D.P.; Serrao, E.; Locke, J.; Swuec, P.; Jónsson, S.R.; Kotecha, A.; Cook, N.J.; Pye, V.E.; Taylor, I.A. A supramolecular assembly mediates lentiviral DNA integration. Science 2017, 355, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.; Hope, T.; Chin, D.; Trono, D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 9825–9830. [Google Scholar] [CrossRef] [PubMed]
- Bouyac-Bertoia, M.; Dvorin, J.D.; Fouchier, R.A.; Jenkins, Y.; Meyer, B.E.; Wu, L.I.; Emerman, M.; Malim, M.H. HIV-1 infection requires a functional integrase NLS. Mol. Cell 2001, 7, 1025–1035. [Google Scholar] [CrossRef]
- Busschots, K.; Voet, A.; De Maeyer, M.; Rain, J.-C.; Emiliani, S.; Benarous, R.; Desender, L.; Debyser, Z.; Christ, F. Identification of the LEDGF/p75 binding site in HIV-1 integrase. J. Mol. Biol. 2007, 365, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Limón, A.; Devroe, E.; Silver, P.A.; Cherepanov, P.; Engelman, A. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 12735–12746. [Google Scholar] [CrossRef] [PubMed]
- Limón, A.; Devroe, E.; Lu, R.; Ghory, H.Z.; Silver, P.A.; Engelman, A. Nuclear localization of human immunodeficiency virus type 1 preintegration complexes (PICs): V165A and R166A are pleiotropic integrase mutants primarily defective for integration, not PIC nuclear import. J. Virol. 2002, 76, 10598–10607. [Google Scholar] [CrossRef] [PubMed]
- Borrenberghs, D.; Dirix, L.; De Wit, F.; Rocha, S.; Blokken, J.; De Houwer, S.; Gijsbers, R.; Christ, F.; Hofkens, J.; Hendrix, J. Dynamic oligomerization of integrase orchestrates HIV nuclear entry. Sci. Rep. 2016, 6, 36485. [Google Scholar] [CrossRef] [PubMed]
- Taltynov, O.; Demeulemeester, J.; Christ, F.; De Houwer, S.; Tsirkone, V.G.; Gerard, M.; Weeks, S.D.; Strelkov, S.V.; Debyser, Z. Interaction of transportin-SR2 with Ras-related nuclear protein (Ran) GTPase. J. Biol. Chem. 2013, 288, 25603–25613. [Google Scholar] [CrossRef] [PubMed]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Rocha, S.; Dirix, L.; Gijsbers, R.; Christ, F.; Debyser, Z. The HIV-1 integrase mutant R263A/K264A is 2-fold defective for TRN-SR2 binding and viral nuclear import. J. Biol. Chem. 2014, 289, 25351–25361. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef]
- Bochnakian, A.; Zhen, A.; Zisoulis, D.G.; Idica, A.; KewalRamani, V.N.; Neel, N.; Daugaard, I.; Hamdorf, M.; Kitchen, S.; Lee, K. Interferon-inducible microRNA miR-128 modulates HIV-1 replication by targeting TNPO3 mRNA. J. Virol. 2019, 93, e00364-19. [Google Scholar] [CrossRef]
- Demeulemeester, J.; Blokken, J.; De Houwer, S.; Dirix, L.; Klaassen, H.; Marchand, A.; Chaltin, P.; Christ, F.; Debyser, Z. Inhibitors of the integrase–transportin-SR2 interaction block HIV nuclear import. Retrovirology 2018, 15, 5. [Google Scholar] [CrossRef]
- Hamid, F.B.; Kim, J.; Shin, C.-G. Characterization of prototype foamy virus infectivity in transportin 3 knockdown human 293t cell line. J. Microbiol. Biotechnol. 2017, 27, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, K.M.; Headey, S.; Telwatte, S.; Tyssen, D.; Hearps, A.C.; Thomas, D.R.; Tachedjian, G.; Jans, D.A. Molecular dissection of an inhibitor targeting the HIV integrase dependent preintegration complex nuclear import. Cell. Microbiol. 2019, 21, e12953. [Google Scholar] [CrossRef] [PubMed]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Taltynov, O.; Zmajkovicova, K.; Christ, F.; Debyser, Z. Identification of residues in the C-terminal domain of HIV-1 integrase that mediate binding to the transportin-SR2 protein. J. Biol. Chem. 2012, 287, 34059–34068. [Google Scholar] [CrossRef] [PubMed]
- Larue, R.; Gupta, K.; Wuensch, C.; Shkriabai, N.; Kessl, J.J.; Danhart, E.; Feng, L.; Taltynov, O.; Christ, F.; Van Duyne, G.D. Interaction of the HIV-1 intasome with transportin 3 protein (TNPO3 or TRN-SR2). J. Biol. Chem. 2012, 287, 34044–34058. [Google Scholar] [CrossRef] [PubMed]
- Thys, W.; De Houwer, S.; Demeulemeester, J.; Taltynov, O.; Vancraenenbroeck, R.; Gérard, M.; De Rijck, J.; Gijsbers, R.; Christ, F.; Debyser, Z. Interplay between HIV entry and transportin-SR2 dependency. Retrovirology 2011, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Erkmann, J.A.; Wagner, E.J.; Dong, J.; Zhang, Y.; Kutay, U.; Marzluff, W.F. Nuclear import of the stem–loop binding protein and localization during the cell cycle. Mol. Biol. Cell 2005, 16, 2960–2971. [Google Scholar] [CrossRef]
- Maris, C.; Dominguez, C.; Allain, F.H.T. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.T.; Patton, J.G. An RNA recognition motif (RRM) is required for the localization of PTB-associated splicing factor (PSF) to subnuclear speckles. Exp. Cell. Res. 2001, 263, 131–144. [Google Scholar] [CrossRef]
- Cribier, A.; Ségéral, E.; Delelis, O.; Parissi, V.; Simon, A.; Ruff, M.; Benarous, R.; Emiliani, S. Mutations affecting interaction of integrase with TNPO3 do not prevent HIV-1 cDNA nuclear import. Retrovirology 2011, 8, 1–14. [Google Scholar] [CrossRef]
- Tsirkone, V.G.; Blokken, J.; De Wit, F.; Breemans, J.; De Houwer, S.; Debyser, Z.; Christ, F.; Strelkov, S.V. N-terminal half of transportin SR2 interacts with HIV integrase. J. Biol. Chem. 2017, 292, 9699–9710. [Google Scholar] [CrossRef] [PubMed]
- Rice, B.L.; Stake, M.S.; Parent, L.J. TNPO3-mediated nuclear entry of the Rous sarcoma virus Gag protein is independent of the cargo-binding domain. J. Virol. 2020, 94, e00640-20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sokolskaja, E.; Jolly, C.; James, W.; Cowley, S.A.; Fassati, A. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 2011, 7, e1002194. [Google Scholar] [CrossRef] [PubMed]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef] [PubMed]
- Melia, M.J.; Kubota, A.; Ortolano, S.; Vílchez, J.J.; Gámez, J.; Tanji, K.; Bonilla, E.; Palenzuela, L.; Fernández-Cadenas, I.; Přistoupilová, A. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain 2013, 136, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mora, S.; De Wit, F.; Garcia-Perez, J.; Bermejo, M.; Lopez-Huertas, M.R.; Mateos, E.; Marti, P.; Rocha, S.; Vigon, L.; Christ, F. The mutation of Transportin 3 gene that causes limb girdle muscular dystrophy 1F induces protection against HIV-1 infection. PLoS Pathog. 2019, 15, e1007958. [Google Scholar] [CrossRef]
- Francis, A.C.; Melikyan, G.B. Live-cell imaging of early steps of single HIV-1 infection. Viruses 2018, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Luban, J. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 2011, 8, 1–19. [Google Scholar] [CrossRef]
- Yamashita, M.; Emerman, M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 2004, 78, 5670–5678. [Google Scholar] [CrossRef]
- Yamashita, M.; Perez, O.; Hope, T.J.; Emerman, M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 2007, 3, e156. [Google Scholar] [CrossRef]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.; Luban, J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 2013, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Di Nunzio, F.; Yang, Y.; Reszka, N.; Lienlaf, M.; Arhel, N.; Perez, P.; Brass, A.L.; Diaz-Griffero, F. TNPO3 is required for HIV-1 replication after nuclear import but prior to integration and binds the HIV-1 core. J. Virol. 2012, 86, 5931–5936. [Google Scholar] [CrossRef] [PubMed]
- Ocwieja, K.E.; Brady, T.L.; Ronen, K.; Huegel, A.; Roth, S.L.; Schaller, T.; James, L.C.; Towers, G.J.; Young, J.A.; Chanda, S.K. HIV integration targeting: A pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog 2011, 7, e1001313. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wilk, T.; Welker, R.; Kräusslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Von Appen, A.; Kosinski, J.; Sparks, L.; Ori, A.; DiGuilio, A.L.; Vollmer, B.; Mackmull, M.-T.; Banterle, N.; Parca, L.; Kastritis, P. In situ structural analysis of the human nuclear pore complex. Nature 2015, 526, 140–143. [Google Scholar] [CrossRef]
- Hulme, A.E.; Kelley, Z.; Foley, D.; Hope, T.J. Complementary assays reveal a low level of CA associated with viral complexes in the nuclei of HIV-1-infected cells. J. Virol. 2015, 89, 5350–5361. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, I.Z.; Dirix, L.; Lemmens, V.; Borrenberghs, D.; De Wit, F.; Vernaillen, F.; Rocha, S.; Christ, F.; Hendrix, J.; Hofkens, J. Capsid-labelled HIV to investigate the role of capsid during nuclear import and integration. J. Virol. 2020, 94, e01024-19. [Google Scholar]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 imaging reveals progression of infection through CA-dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef]
- Hulme, A.E.; Kelley, Z.; Okocha, E.A.; Hope, T.J. Identification of capsid mutations that alter the rate of HIV-1 uncoating in infected cells. J. Virol. 2015, 89, 643–651. [Google Scholar] [CrossRef]
- Ingram, Z.; Taylor, M.; Okland, G.; Martin, R.; Hulme, A.E. Characterization of HIV-1 uncoating in human microglial cell lines. Virol. J. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the core leads HIV-1 preintegration complex into the nucleus of human lymphocytes. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Zila, V.; Margiotta, E.; Turonova, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Marin, M.; Prellberg, M.J.; Palermino-Rowland, K.; Melikyan, G.B. HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages. Viruses 2020, 12, 1234. [Google Scholar] [CrossRef]
- Rensen, E.; Mueller, F.; Scoca, V.; Parmar, J.J.; Souque, P.; Zimmer, C.; Di Nunzio, F. Clustering and reverse transcription of HIV-1 genomes in nuclear niches of macrophages. EMBO J. 2021, 40, e105247. [Google Scholar] [CrossRef] [PubMed]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, M.-D.; Aringhieri, C.; Vivarelli, S.; Cardinale, S.; Paro, S.; Schümperli, D.; Barabino, S.M. Mammalian pre-mRNA 3′ end processing factor CF Im68 functions in mRNA export. Mol. Biol. Cell 2009, 20, 5211–5223. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ning, J.; Boggs, E.A.; Jang, S.; Wallace, C.; Telmer, C.; Bruchez, M.; Ahn, J.; Engelman, A.; Watkins, S.C. Cytoplasmic CPSF6 regulates HIV-1 capsid trafficking and infection in a cyclophilin A-dependent manner. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Börner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe 2018, 24, 392–404.e398. [Google Scholar] [CrossRef] [PubMed]
- Logue, E.C.; Taylor, K.T.; Goff, P.H.; Landau, N.R. The cargo-binding domain of transportin 3 is required for lentivirus nuclear import. J. Virol. 2011, 85, 12950–12961. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; Valle-Casuso, J.C.; White, T.E.; Brandariz-Nuñez, A.; Bosche, W.J.; Reszka, N.; Gorelick, R.; Diaz-Griffero, F. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 2013, 10, 1–14. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 resistance to the capsid-targeting inhibitor PF74 results in altered dependence on host factors required for virus nuclear entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef]
- Zila, V.; Müller, T.G.; Laketa, V.; Müller, B.; Kräusslich, H.-G. Analysis of CA content and CPSF6 dependence of early HIV-1 replication complexes in SupT1-R5 cells. MBio 2019, 10. [Google Scholar] [CrossRef]
- Scoca, V.; Louveaux, M.; Morin, R.; Ershov, D.; Tinevez, J.-Y.; Di Nunzio, F. Direct tracking of single proviruses reveals HIV-1/LEDGF complexes excluded from virus-induced membraneless organelles. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bonnon, C.; Wendeler, M.W.; Paccaud, J.-P.; Hauri, H.-P. Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J. Cell Sci. 2010, 123, 1705–1715. [Google Scholar] [CrossRef]
- Rebensburg, S.V.; Wei, G.; Larue, R.C.; Lindenberger, J.; Francis, A.C.; Annamalai, A.S.; Morrison, J.; Shkriabai, N.; Huang, S.-W.; KewalRamani, V. Sec24C is an HIV-1 host dependency factor crucial for virus replication. Nat. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Lemke, C.T.; Titolo, S.; von Schwedler, U.; Goudreau, N.; Mercier, J.-F.; Wardrop, E.; Faucher, A.-M.; Coulombe, R.; Banik, S.S.; Fader, L. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 2012, 86, 6643–6655. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; Brandariz-Nuñez, A.; Wang, X.; Smith, A.B.; Diaz-Griffero, F. Human cytosolic extracts stabilize the HIV-1 core. J. Virol. 2013, 87, 10587–10597. [Google Scholar] [CrossRef]
- Lad, L.; Clancy, S.; Koditek, D.; Wong, M.H.; Jin, D.; Niedziela-Majka, A.; Papalia, G.A.; Hung, M.; Yant, S.; Somoza, J.R. Functional label-free assays for characterizing the in vitro mechanism of action of small molecule modulators of capsid assembly. Biochemistry 2015, 54, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Ferhadian, D.; Sowd, G.A.; Serrao, E.; Shi, J.; Halambage, U.D.; Teng, S.; Soto, J.; Siddiqui, M.A.; Engelman, A.N. Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74. J. Virol. 2016, 90, 5808–5823. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 reinforces the HIV-1 capsid to impair reverse transcription-induced uncoating. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Link, J.O.; Rhee, M.S.; Winston, C.T.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 1–6. [Google Scholar]
- Goldstone, D.C.; Yap, M.W.; Robertson, L.E.; Haire, L.F.; Taylor, W.R.; Katzourakis, A.; Stoye, J.P.; Taylor, I.A. Structural and functional analysis of prehistoric lentiviruses uncovers an ancient molecular interface. Cell Host Microbe 2010, 8, 248–259. [Google Scholar] [CrossRef]
- Yap, M.W.; Dodding, M.P.; Stoye, J.P. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 2006, 80, 4061–4067. [Google Scholar] [CrossRef] [PubMed]
- Briones, M.S.; Dobard, C.W.; Chow, S.A. Role of human immunodeficiency virus type 1 integrase in uncoating of the viral core. J. Virol. 2010, 84, 5181–5190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 1996, 70, 3551–3560. [Google Scholar] [CrossRef]
- Braaten, D.; Luban, J. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001, 20, 1300–1309. [Google Scholar] [CrossRef]
- De Iaco, A.; Luban, J. Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.B.; Shi, J.; Hout, D.R.; Oztop, I.; Krishnan, L.; Ahn, J.; Shotwell, M.S.; Engelman, A.; Aiken, C. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J. Virol. 2013, 87, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z. Cytopmasic CPSF6 and Cyclophilin A Modulate HIV-1 Trafficking; University of Pittsburgh: Pittsburgh, PA, USA, 2020. [Google Scholar]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Di Nunzio, F.; Zimmer, C. FlAsH-PALM: Super-Resolution Pointillist Imaging with FlAsH-Tetracysteine Labeling. In Exocytosis and Endocytosis; Springer: Berlin/Heidelberg, Germany, 2014; pp. 183–193. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabasi, M.; Nombela, I.; Janssens, J.; Lahousse, A.P.; Christ, F.; Debyser, Z. Role of Transportin-SR2 in HIV-1 Nuclear Import. Viruses 2021, 13, 829. https://doi.org/10.3390/v13050829
Tabasi M, Nombela I, Janssens J, Lahousse AP, Christ F, Debyser Z. Role of Transportin-SR2 in HIV-1 Nuclear Import. Viruses. 2021; 13(5):829. https://doi.org/10.3390/v13050829
Chicago/Turabian StyleTabasi, Maryam, Ivan Nombela, Julie Janssens, Adrien P. Lahousse, Frauke Christ, and Zeger Debyser. 2021. "Role of Transportin-SR2 in HIV-1 Nuclear Import" Viruses 13, no. 5: 829. https://doi.org/10.3390/v13050829
APA StyleTabasi, M., Nombela, I., Janssens, J., Lahousse, A. P., Christ, F., & Debyser, Z. (2021). Role of Transportin-SR2 in HIV-1 Nuclear Import. Viruses, 13(5), 829. https://doi.org/10.3390/v13050829