
Complement Proteins as Soluble Pattern Recognition Receptors for Pathogenic Viruses


 ,
, 
Abstract
1. Introduction
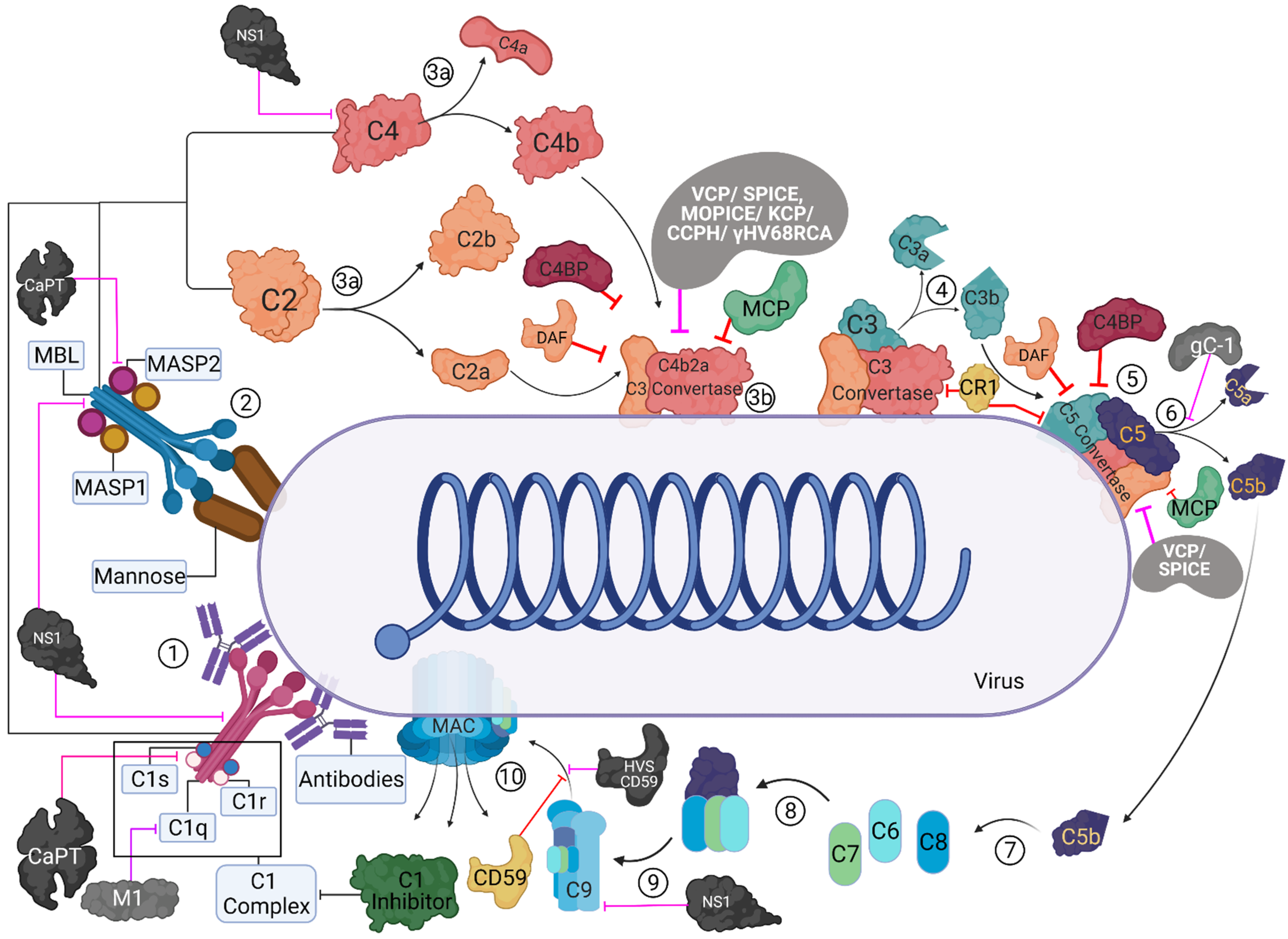
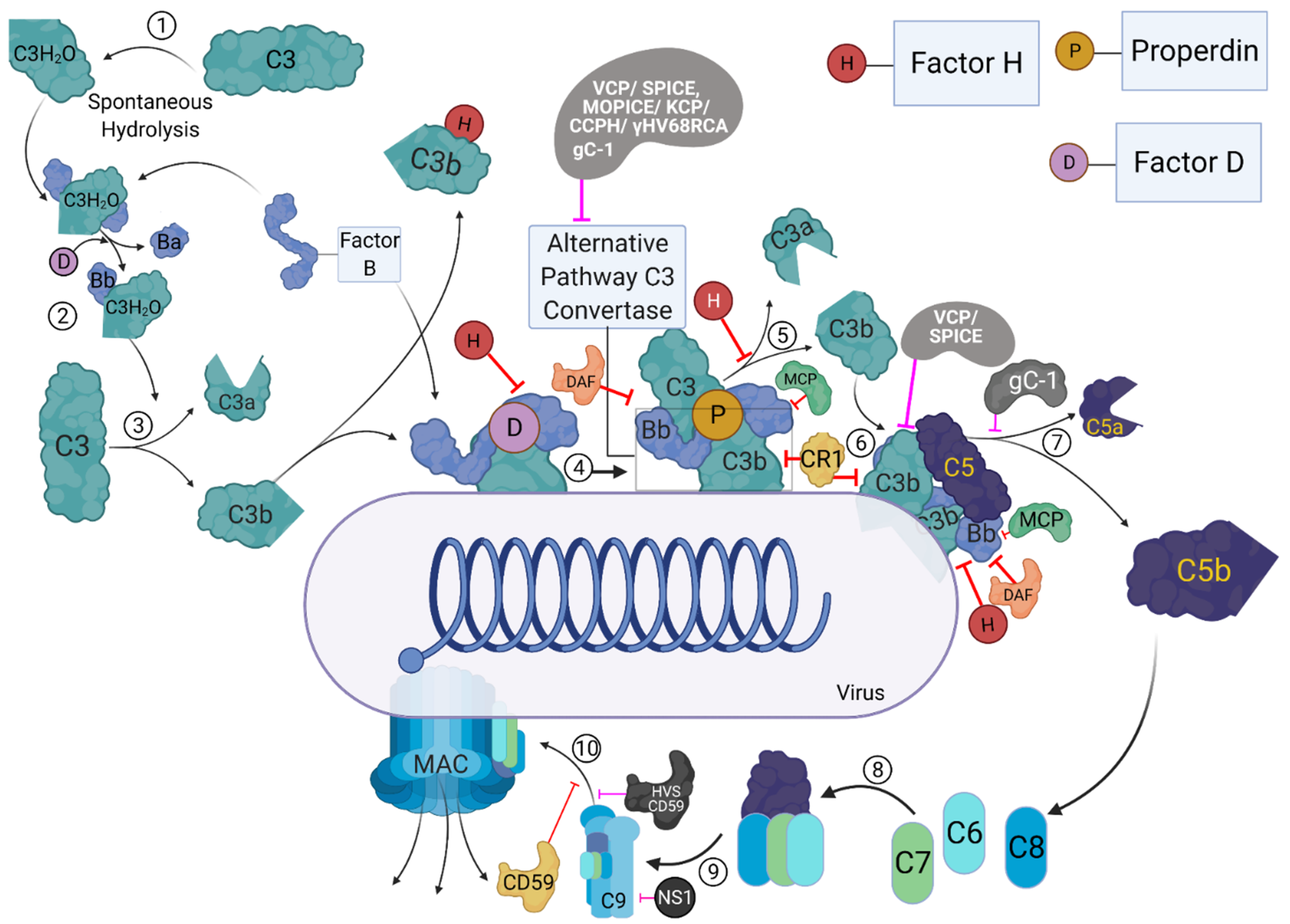
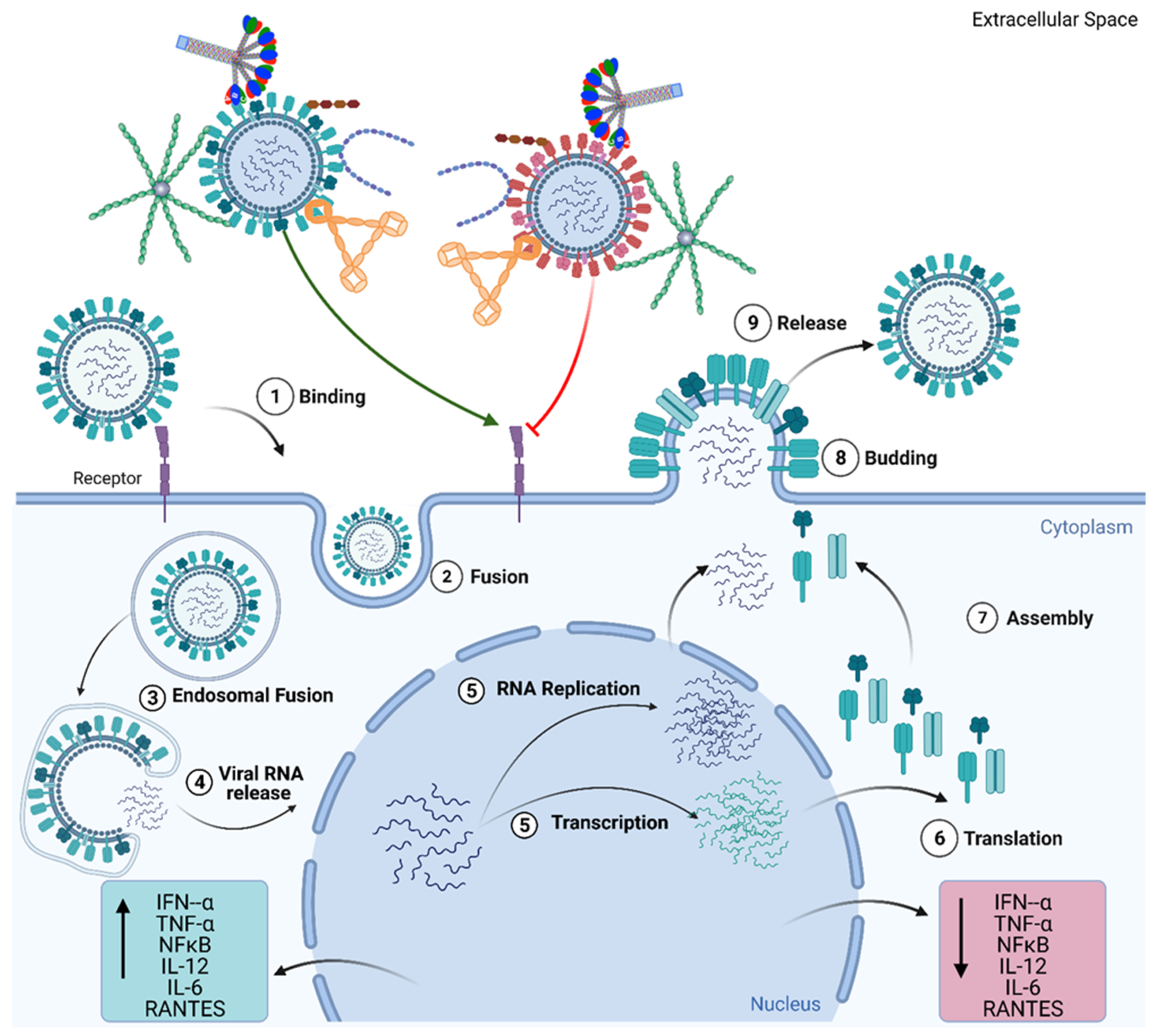
2. Role of the Complement System during Viral Infection: Viral Evasion Mechanisms
3. C1q Exploiting Viral Evasion Mechanisms
4. Viral Evasion Strategies Exploiting C4b Binding Protein
5. Involvement of Properdin in Anti-Viral Immune Response and Viral Evasion
6. Factor H
7. Human Mannan-Binding Lectin (MBL)
8. Ficolins
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sim, R.B.; Tsiftsoglou, S.A. Proteases of the complement system. Biochem. Soc. Trans. 2004, 32, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Müller-Eberhard, H.J.; Polley, M.J.; Calcott, M.A. Formation and Functional Significance of a Molecular Complex Derived from the Second and the Fourth Component of Human Complement. J. Exp. Med. 1967, 125, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Naff, G.B.; Pensky, J.; Lepow, I.H. The Macromolecular Nature of the First Component of Human Complement. J. Exp. Med. 1964, 119, 593–613. [Google Scholar] [CrossRef]
- Polley, M.J.; Müller-Eberhard, H.J. The second component of human complement: Its isolation, fragmentation by C’1 esterase, and incorporation into C’3 convertase. J. Exp. Med. 1968, 128, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Kishore, U.; Reid, K.B. Modular organization of proteins containing C1q-like globular domain. Immunopharmacology 1999, 42, 15–21. [Google Scholar] [CrossRef]
- Kouser, L.; Abdul-Aziz, M.; Nayak, A.; Stover, C.M.; Sim, R.B.; Kishore, U. Properdin and factor h: Opposing players on the alternative complement pathway “see-saw”. Front Immunol. 2013, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Bouwman, L.H.; van Gijlswijk-Janssen, D.J.; Faber-Krol, M.C.; Stahl, G.L.; Daha, M.R. Human IgA Activates the Complement System Via the Mannan-Binding Lectin Pathway. J. Immunol. 2001, 167, 2861–2868. [Google Scholar] [CrossRef]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation changes of IgG associated with rheumatooid arthritis can activate complement via the mannose-binding protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef]
- Lu, J.; Kishore, U. C1 Complex: An Adaptable Proteolytic Module for Complement and Non-Complement Functions. Front Immunol. 2017, 8, 592. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5–C5a receptor axis. Mol. Immunol. 2011, 48, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Muller-Eberhard, H.J. The Membrane Attack Complex of Complement. Annu. Rev. Immunol. 1986, 4, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 2009, 41, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; McGeer, P.L. Physiology and Pathophysiology of Complement: Progress and Trends. Crit. Rev. Clin. Lab. Sci. 1995, 32, 265–298. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Ember, J.; Jagels, M.; Hugli, T. Characterization of complement anaphylatoxins and their biological responses. Hum. Complement Syst. Health Dis. 1998, 241. [Google Scholar] [CrossRef]
- Murakami, Y.; Imamichi, T.; Nagasawa, S. Characterization of C3a anaphylatoxin receptor on guinea-pig macrophages. Immunology 1993, 79, 633–638. [Google Scholar]
- Elsner, J.; Oppermann, M.; Czech, W.; Kapp, A. C3a activates the respiratory burst in human polymorphonuclear neutrophilic leukocytes via pertussis toxin-sensitive G-proteins. Blood 1994, 83, 3324–3331. [Google Scholar] [CrossRef]
- Elsner, J.; Oppermann, M.; Czech, W.; Dobos, G.; Schöpf, E.; Norgauer, J.; Kapp, A. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur. J. Immunol. 1994, 24, 518–522. [Google Scholar] [CrossRef]
- El-Lati, S.G.; Church, M.K.; Dahinden, C.A. Complement Peptides C3a- and C5a-Induced Mediator Release from Dissociated Human Skin Mast Cells. J. Investig. Dermatol. 1994, 102, 803–806. [Google Scholar] [CrossRef]
- Kretzschmar, T.; Jeromin, A.; Gietz, C.; Bautsch, W.; Klos, A.; Köhl, J.; Rechkemmer, G.; Bitter-Suermann, D. Chronic myelogenous leukemia-derived basophilic granulocytes express a functional active receptor for the anaphylatoxin C3a. Eur. J. Immunol. 1993, 23, 558–561. [Google Scholar] [CrossRef]
- Hartmann, K.; Henz, B.M.; Krüger-Krasagakes, S.; Köhl, J.; Burger, R.; Guhl, S.; Haase, I.; Lippert, U.; Zuberbier, T. C3a and C5a Stimulate Chemotaxis of Human Mast Cells. Blood 1997, 89, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.H.; Jagels, M.A.; Hugli, T.E. Regulation of IL-6 Synthesis in Human Peripheral Blood Mononuclear Cells by C3a and C3adesArg. J. Immunol. 1999, 162, 453–459. [Google Scholar]
- Fischer, W.H.; Hugli, T.E. Regulation of B cell functions by C3a and C3a(desArg): Suppression of TNF-alpha, IL-6, and the polyclonal immune response. J. Immunol. 1997, 159, 4279–4286. [Google Scholar] [PubMed]
- Peng, Q.; Li, K.; Sacks, S.H.; Zhou, W. The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm. Allergy Drug Targets 2009, 8, 236–246. [Google Scholar] [CrossRef]
- Gorsuch, W.B.; Chrysanthou, E.; Schwaeble, W.J.; Stahl, G.L. The complement system in ischemia–reperfusion injuries. Immunobiology 2012, 217, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Farrar, C.; Asgari, E.; Schwaeble, W.; Sacks, S. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front. Immunol. 2012, 3. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- Janeway, C. Immunobiology: The Immune System in Health and Disease, 4th ed.; Current Biology Publications; Garland Pub.: London, UK; New York, NY, USA, 1999; p. xix. 635p. [Google Scholar]
- Chen, J.Y.; Cortes, C.; Ferreira, V.P. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol. Immunol. 2018, 102, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Pangburn, M.K.; Cortes, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Sim, R.B.; Day, A.J.; Moffatt, B.E.; Fontaine, M. Complement factor I and cofactors in control of complement system convertase enzymes. Methods Enzym. 1993, 223, 13–35. [Google Scholar] [CrossRef]
- Blom, A.M.; Kask, L.; Dahlbäck, B. Structural Requirements for the Complement Regulatory Activities of C4BP *. J. Biol. Chem. 2001, 276, 27136–27144. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Nussenzweig, V. The role of C4-binding protein and {beta }1H in proteolysis of C4b and C3b. J. Exp. Med. 1979, 150, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Tamura, N. Interaction of C4-binding protein with cell-bound C4b. A quantitative analysis of binding and the role of C4-binding protein in proteolysis of cell-bound C4b. J. Exp. Med. 1983, 157, 1239–1251. [Google Scholar] [CrossRef]
- Lublin, D.M.; Atkinson, J.P. Decay-Accelerating Factor: Biochemistry, Molecular Biology, and Function. Annu. Rev. Immunol. 1989, 7, 35–58. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Post, T.W.; Atkinson, J.P. Membrane Cofactor Protein (MCP or CD46): Newest Member of the Regulators of Complement Activation Gene Cluster. Annu. Rev. Immunol. 1991, 9, 431–455. [Google Scholar] [CrossRef]
- Ahearn, J.M.; Fearon, D.T. Structure and Function of the Complement Receptors, CR1 (CD35) and CR2 (CD21). In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1989; Volume 46, pp. 183–219. [Google Scholar]
- Rollins, S.A.; Sims, P.J. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J. Immunol. 1990, 144, 3478–3483. [Google Scholar]
- Berry, D.M.; Almeida, J.D. The Morphological and Biological Effects of Various Antisera on Avian Infectious Bronchitis Virus. J. Gen. Virol. 1968, 3, 97–102. [Google Scholar] [CrossRef]
- Cooper, N. Complement and viruses. In The Human Complement System in Health and Disease; Marcel DekkerInc.: New York, NY, USA, 1998; pp. 393–407. [Google Scholar]
- Cooper, N.R.; Nemerow, G.R. Complement, viruses, and virus-infected cells. Springer Semin. Immunopathol. 1983, 6, 327–347. [Google Scholar] [CrossRef]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural Antibody and Complement Mediate Neutralization of Influenza Virus in the Absence of Prior Immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef]
- Tam, J.C.H.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular sensing of complement C3 activates cell autonomous immunity. Science 2014, 345, 1256070. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Olinger, G.G.; Aris, S.; Chen, Y.; Gewurz, H.; Spear, G.T. Mannose-binding lectin binds to Ebola and Marburg envelope glycoproteins, resulting in blocking of virus interaction with DC-SIGN and complement-mediated virus neutralization. J. Gen. Virol. 2005, 86, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Blue, C.E.; Spiller, O.B.; Blackbourn, D.J. The relevance of complement to virus biology. Virology 2004, 319, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, A.; Fauquert, J.-L.; Thomas, C.; Kemper, C.; Drouet, C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol. Immunol. 2014, 58, 98–107. [Google Scholar] [CrossRef]
- Weaver, D.J., Jr.; Reis, E.S.; Pandey, M.K.; Köhl, G.; Harris, N.; Gerard, C.; Köhl, J. C5a receptor-deficient dendritic cells promote induction of Treg and Th17 cells. Eur. J. Immunol. 2010, 40, 710–721. [Google Scholar] [CrossRef]
- Barrington, R.A.; Schneider, T.J.; Pitcher, L.A.; Mempel, T.R.; Ma, M.; Barteneva, N.S.; Carroll, M.C. Uncoupling CD21 and CD19 of the B-cell coreceptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14490–14495. [Google Scholar] [CrossRef]
- Fodor, W.L.; Rollins, S.A.; Bianco-Caron, S.; Rother, R.P.; Guilmette, E.R.; Burton, W.V.; Albrecht, J.C.; Fleckenstein, B.; Squinto, S.P. The complement control protein homolog of herpesvirus saimiri regulates serum complement by inhibiting C3 convertase activity. J. Virol. 1995, 69, 3889–3892. [Google Scholar] [CrossRef]
- Singh, A.K.; Mullick, J.; Bernet, J.; Sahu, A. Functional Characterization of the Complement Control Protein Homolog of Herpesvirus Saimiri: ARG-118 IS CRITICAL FOR FACTOR I COFACTOR ACTIVITIES. J. Biol. Chem. 2006, 281, 23119–23128. [Google Scholar] [CrossRef]
- Albrecht, J.C.; Fleckenstein, B. New member of the multigene family of complement control proteins in herpesvirus saimiri. J. Virol. 1992, 66, 3937–3940. [Google Scholar] [CrossRef]
- Mullick, J.; Singh, A.K.; Panse, Y.; Yadav, V.; Bernet, J.; Sahu, A. Identification of Functional Domains in Kaposica, the Complement Control Protein Homolog of Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus 8). J. Virol. 2005, 79, 5850–5856. [Google Scholar] [CrossRef]
- Mullick, J.; Bernet, J.; Singh, A.K.; Lambris, J.D.; Sahu, A. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) open reading frame 4 protein (Kaposica) is a functional homolog of complement control proteins. J. Virol. 2003, 77, 3878–3881. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Blackbourn, D.J.; Mark, L.; Proctor, D.G.; Blom, A.M. Functional Activity of the Complement Regulator Encoded by Kaposi’s Sarcoma-associated Herpesvirus*. J. Biol. Chem. 2003, 278, 9283–9289. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.-C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Molina, H.; van Berkel, V.; Speck, S.H.; Virgin, H.W. Murine Gammaherpesvirus 68 Encodes a Functional Regulator of Complement Activation. J. Virol. 1999, 73, 7658–7670. [Google Scholar] [CrossRef]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Rosengard, A.M.; Liu, Y.; Nie, Z.; Jimenez, R. Variola virus immune evasion design: Expression of a highly efficient inhibitor of human complement. Proc. Natl. Acad. Sci. USA 2002, 99, 8808–8813. [Google Scholar] [CrossRef]
- Kotwal, G.J.; Moss, B. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature 1988, 335, 176–178. [Google Scholar] [CrossRef]
- Kotwal, G.; Isaacs, S.; McKenzie, R.; Frank, M.; Moss, B. Inhibition of the complement cascade by the major secretory protein of vaccinia virus. Science 1990, 250, 827–830. [Google Scholar] [CrossRef]
- Sahu, A.; Isaacs, S.N.; Soulika, A.M.; Lambris, J.D. Interaction of Vaccinia Virus Complement Control Protein with Human Complement Proteins: Factor I-Mediated Degradation of C3b to iC3b1 Inactivates the Alternative Complement Pathway. J. Immunol. 1998, 160, 5596–5604. [Google Scholar] [CrossRef]
- McKenzie, R.; Kotwal, G.J.; Moss, B.; Hammer, C.H.; Frank, M.M. Regulation of Complement Activity by Vaccinia Virus Complement-Control Protein. J. Infect. Dis. 1992, 166, 1245–1250. [Google Scholar] [CrossRef]
- Vanderplasschen, A.; Mathew, E.; Hollinshead, M.; Sim, R.B.; Smith, G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. USA 1998, 95, 7544–7549. [Google Scholar] [CrossRef]
- Bonaparte, R.S.; Hair, P.S.; Banthia, D.; Marshall, D.M.; Cunnion, K.M.; Krishna, N.K. Human Astrovirus Coat Protein Inhibits Serum Complement Activation via C1, the First Component of the Classical Pathway. J. Virol. 2008, 82, 817–827. [Google Scholar] [CrossRef]
- Hair, P.S.; Gronemus, J.Q.; Crawford, K.B.; Salvi, V.P.; Cunnion, K.M.; Thielens, N.M.; Arlaud, G.J.; Rawal, N.; Krishna, N.K. Human astrovirus coat protein binds C1q and MBL and inhibits the classical and lectin pathways of complement activation. Mol. Immunol. 2010, 47, 792–798. [Google Scholar] [CrossRef]
- Gronemus, J.Q.; Hair, P.S.; Crawford, K.B.; Nyalwidhe, J.O.; Cunnion, K.M.; Krishna, N.K. Potent inhibition of the classical pathway of complement by a novel C1q-binding peptide derived from the human astrovirus coat protein. Mol. Immunol. 2010, 48, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.; Wang, L.; Mastellos, D.; Sahu, A.; Lambris, J.D.; Friedman, H.M. In Vivo Role of Complement-Interacting Domains of Herpes Simplex Virus Type 1 Glycoprotein Gc. J. Exp. Med. 1999, 190, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-L.; Peng, C.; Kostavasili, I.; Friedman, H.M.; Lambris, J.D.; Eisenberg, R.J.; Cohen, G.H. The interaction of glycoprotein C of herpes simplex virus types 1 and 2 with the alternative complement pathway. Virology 1994, 203, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Kostavasili, I.; Sahu, A.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H.; Lambris, J.D. Mechanism of complement inactivation by glycoprotein C of herpes simplex virus. J. Immunol. 1997, 158, 1763–1771. [Google Scholar] [PubMed]
- Fries, L.F.; Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Hammer, C.H.; Frank, M.M. Glycoprotein C of herpes simplex virus 1 is an inhibitor of the complement cascade. J. Immunol. 1986, 137, 1636–1641. [Google Scholar] [PubMed]
- Harris, S.L.; Frank, I.; Vee, A.; Cohen, G.H.; Eisenberg, R.J.; Friedman, H.M. Glycoprotein C of Herpes Simplex Virus Type 1 Prevents Complement-Mediated Cell Lysis and Virus Neutralization. J. Infect. Dis. 1990, 162, 331–337. [Google Scholar] [CrossRef]
- Rux, A.H.; Lou, H.; Lambris, J.D.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H. Kinetic Analysis of Glycoprotein C of Herpes Simplex Virus Types 1 and 2 Binding to Heparin, Heparan Sulfate, and Complement Component C3b. Virology 2002, 294, 324–332. [Google Scholar] [CrossRef][Green Version]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, J.J.; Brandriss, M.W.; Putnak, J.R.; Walsh, E.E. Cell surface expression of yellow fever virus non-structural glycoprotein NS1: Consequences of interaction with antibody. J. Gen. Virol. 1990, 71, 593–599. [Google Scholar] [CrossRef]
- Winkler, G.; Maxwell, S.E.; Ruemmler, C.; Stollar, V. Newly synthesized dengue-2 virus nonstructural protein NS1 is a soluble protein but becomes partially hydrophobic and membrane-associated after dimerization. Virology 1989, 171, 302–305. [Google Scholar] [CrossRef]
- Avirutnan, P.; Fuchs, A.; Hauhart, R.E.; Somnuke, P.; Youn, S.; Diamond, M.S.; Atkinson, J.P. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J. Exp. Med. 2010, 207, 793–806. [Google Scholar] [CrossRef]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of Flavivirus Nonstructural Protein NS1 to C4b Binding Protein Modulates Complement Activation. J. Immunol. 2011, 187, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, T.; Chaichana, P.; Yamate, M.; Anantapreecha, S.; Ikuta, K. Secreted complement regulatory protein clusterin interacts with dengue virus nonstructural protein 1. Biochem. Biophys. Res. Commun. 2007, 362, 1051–1056. [Google Scholar] [CrossRef]
- Conde, J.N.; da Silva, E.M.; Allonso, D.; Coelho, D.R.; Andrade, I.d.S.; de Medeiros, L.N.; Menezes, J.L.; Barbosa, A.S.; Mohana-Borges, R. Inhibition of the Membrane Attack Complex by Dengue Virus NS1 through Interaction with Vitronectin and Terminal Complement Proteins. J. Virol. 2016, 90, 9570–9581. [Google Scholar] [CrossRef]
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Jensenius, J.C.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl. Acad. Sci. USA 2017, 114, 986–991. [Google Scholar] [CrossRef]
- Kishore, U.; Reid, K.B. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Reis, E.S.; Barbuto, J.A.; Isaac, L. Complement components, regulators and receptors are produced by human monocyte-derived dendritic cells. Immunobiology 2007, 212, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Woltman, A.M.; Nauta, A.J.; Roos, A.; Trouw, L.A.; Seelen, M.A.; Schena, F.P.; Daha, M.R.; van Kooten, C. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood 2004, 103, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Loos, M. Expression of membrane C1q in human monocyte-derived macrophages is developmentally regulated and enhanced by interferon-gamma. FEBS Lett. 2001, 500, 91–98. [Google Scholar] [CrossRef]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 10346. [Google Scholar] [CrossRef]
- Mozdzanowska, K.; Feng, J.; Eid, M.; Zharikova, D.; Gerhard, W. Enhancement of neutralizing activity of influenza virus-specific antibodies by serum components. Virology 2006, 352, 418–426. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758. [Google Scholar] [CrossRef]
- Thielens, N.M.; Tacnet-Delorme, P.; Arlaud, G.J. Interaction of C1q and mannan-binding lectin with viruses. Immunobiology 2002, 205, 563–574. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Vieillard, V.; Sagan, S.; Bismuth, G.; Debre, P. HIV gp41 engages gC1qR on CD4+ T cells to induce the expression of an NK ligand through the PIP3/H2O2 pathway. PLoS Pathog. 2010, 6, e1000975. [Google Scholar] [CrossRef]
- Ebenbichler, C.F.; Thielens, N.M.; Vornhagen, R.; Marschang, P.; Arlaud, G.J.; Dierich, M.P. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J. Exp. Med. 1991, 174, 1417–1424. [Google Scholar] [CrossRef]
- Thielens, N.M.; Bally, I.M.; Ebenbichler, C.F.; Dierich, M.P.; Arlaud, G.J. Further characterization of the interaction between the C1q subcomponent of human C1 and the transmembrane envelope glycoprotein gp41 of HIV-1. J. Immunol. 1993, 151, 6583–6592. [Google Scholar]
- Kishore, U.; Gupta, S.K.; Perdikoulis, M.V.; Kojouharova, M.S.; Urban, B.C.; Reid, K.B. Modular organization of the carboxyl-terminal, globular head region of human C1q A, B, and C chains. J. Immunol. 2003, 171, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Pinter, A.; Honnen, W.J.; Tilley, S.A.; Bona, C.; Zaghouani, H.; Gorny, M.K.; Zolla-Pazner, S. Oligomeric structure of gp41, the transmembrane protein of human immunodeficiency virus type 1. J. Virol. 1989, 63, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Marschang, P.; Kruger, U.; Ochsenbauer, C.; Gurtler, L.; Hittmair, A.; Bosch, V.; Patsch, J.R.; Dierich, M.P. Complement activation by HIV-1-infected cells: The role of transmembrane glycoprotein gp41. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 14, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, H.; Ebenbichler, C.; Schneider, R.; Janatova, J.; Dierich, M.P. Interaction of several complement proteins with gp120 and gp41, the two envelope glycoproteins of HIV-1. AIDS 1995, 9, 19–26. [Google Scholar] [CrossRef]
- Stoiber, H.; Thielens, N.M.; Ebenbichler, C.; Arlaud, G.J.; Dierich, M.P. The envelope glycoprotein of HIV-1 gp120 and human complement protein C1q bind to the same peptides derived from three different regions of gp41, the transmembrane glycoprotein of HIV-1, and share antigenic homology. Eur J. Immunol. 1994, 24, 294–300. [Google Scholar] [CrossRef]
- Susal, C.; Kirschfink, M.; Kropelin, M.; Daniel, V.; Opelz, G. Identification of complement activation sites in human immunodeficiency virus type-1 glycoprotein gp120. Blood 1996, 87, 2329–2336. [Google Scholar] [CrossRef]
- Aasa-Chapman, M.M.; Holuigue, S.; Aubin, K.; Wong, M.; Jones, N.A.; Cornforth, D.; Pellegrino, P.; Newton, P.; Williams, I.; Borrow, P.; et al. Detection of antibody-dependent complement-mediated inactivation of both autologous and heterologous virus in primary human immunodeficiency virus type 1 infection. J. Virol. 2005, 79, 2823–2830. [Google Scholar] [CrossRef][Green Version]
- Pednekar, L.; Pandit, H.; Paudyal, B.; Kaur, A.; Al-Mozaini, M.A.; Kouser, L.; Ghebrehiwet, B.; Mitchell, D.A.; Madan, T.; Kishore, U. Complement Protein C1q Interacts with DC-SIGN via Its Globular Domain and Thus May Interfere with HIV-1 Transmission. Front. Immunol. 2016, 7, 600. [Google Scholar] [CrossRef]
- Szabo, J.; Cervenak, L.; Toth, F.D.; Prohaszka, Z.; Horvath, L.; Kerekes, K.; Beck, Z.; Bacsi, A.; Erdei, A.; Peerschke, E.I.; et al. Soluble gC1q-R/p33, a cell protein that binds to the globular “heads” of C1q, effectively inhibits the growth of HIV-1 strains in cell cultures. Clin. Immunol. 2001, 99, 222–231. [Google Scholar] [CrossRef]
- Kittlesen, D.J.; Chianese-Bullock, K.A.; Yao, Z.Q.; Braciale, T.J.; Hahn, Y.S. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J. Clin. Investig. 2000, 106, 1239–1249. [Google Scholar] [CrossRef]
- Mohan, K.V.; Ghebrehiwet, B.; Atreya, C.D. The N-terminal conserved domain of rubella virus capsid interacts with the C-terminal region of cellular p32 and overexpression of p32 enhances the viral infectivity. Virus Res. 2002, 85, 151–161. [Google Scholar] [CrossRef]
- Matthews, D.A.; Russell, W.C. Adenovirus core protein V interacts with p32--a protein which is associated with both the mitochondria and the nucleus. J. Gen. Virol. 1998, 79 Pt 7, 1677–1685. [Google Scholar] [CrossRef]
- Wang, Y.; Finan, J.E.; Middeldorp, J.M.; Hayward, S.D. P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein-Barr virus. Virology 1997, 236, 18–29. [Google Scholar] [CrossRef][Green Version]
- Vieillard, V.; Strominger, J.L.; Debre, P. NK cytotoxicity against CD4+ T cells during HIV-1 infection: A gp41 peptide induces the expression of an NKp44 ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 10981–10986. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.; Ghebrehiwet, B.; Cossart, P. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. EMBO J. 2000, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Spear, G.T.; Jiang, H.X.; Sullivan, B.L.; Gewurz, H.; Landay, A.L.; Lint, T.F. Direct binding of complement component C1q to human immunodeficiency virus (HIV) and human T lymphotrophic virus-I (HTLV-I) coinfected cells. AIDS Res. Hum. Retrovir. 1991, 7, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Haraguchi, Y.; Jinno, A.; Iino, Y.; Morishita, Y.; Shiraki, H.; Hoshino, H. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J. Immunol. 1998, 161, 5712–5719. [Google Scholar] [PubMed]
- Sagara, Y.; Inoue, Y.; Shiraki, H.; Jinno, A.; Hoshino, H.; Maeda, Y. Identification and mapping of functional domains on human T-cell lymphotropic virus type 1 envelope proteins by using synthetic peptides. J. Virol. 1996, 70, 1564–1569. [Google Scholar] [CrossRef]
- Bartholomew, R.M.; Esser, A.F.; Muller-Eberhard, H.J. Lysis of oncornaviruses by human serum. Isolation of the viral complement (C1) receptor and identification as p15E. J. Exp. Med. 1978, 147, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Kunnakkadan, U.; Nag, J.; Kumar, N.A.; Mukesh, R.K.; Suma, S.M.; Johnson, J.B. Complement-Mediated Neutralization of a Potent Neurotropic Human Pathogen, Chandipura Virus, Is Dependent on C1q. J. Virol. 2019, 93, e00994-19. [Google Scholar] [CrossRef]
- Blom, A.M. Structural and functional studies of complement inhibitor C4b-binding protein. Biochem. Soc. Trans. 2002, 30, 978–982. [Google Scholar] [CrossRef]
- Norman, D.G.; Barlow, P.N.; Baron, M.; Day, A.J.; Sim, R.B.; Campbell, I.D. Three-dimensional structure of a complement control protein module in solution. J. Mol. Biol. 1991, 219, 717–725. [Google Scholar] [CrossRef]
- Blom, A.M.; Villoutreix, B.O.; Dahlbäck, B. Mutations in α-Chain of C4BP That Selectively Affect Its Factor I Cofactor Function*. J. Biol. Chem. 2003, 278, 43437–43442. [Google Scholar] [CrossRef]
- Blom, A.M.; Webb, J.; Villoutreix, B.O.; Dahlbäck, B. A Cluster of Positively Charged Amino Acids in the C4BP α-Chain Is Crucial for C4b Binding and Factor I Cofactor Function*. J. Biol. Chem. 1999, 274, 19237–19245. [Google Scholar] [CrossRef]
- Feng, G.; Li, J.; Zheng, M.; Yang, Z.; Liu, Y.; Zhang, S.; Ye, L.; Zhang, W.; Zhang, X. Hepatitis B virus X protein up-regulates C4b-binding protein α through activating transcription factor Sp1 in protection of hepatoma cells from complement attack. Oncotarget 2016, 7, 28013–28026. [Google Scholar] [CrossRef]
- Shan, C.; Zhang, S.; Cui, W.; You, X.; Kong, G.; Du, Y.; Qiu, L.; Ye, L.; Zhang, X. Hepatitis B virus X protein activates CD59 involving DNA binding and let-7i in protection of hepatoma and hepatic cells from complement attack. Carcinogenesis 2011, 32, 1190–1197. [Google Scholar] [CrossRef][Green Version]
- Zhang, S.; Shan, C.; Cui, W.; You, X.; Du, Y.; Kong, G.; Gao, F.; Ye, L.; Zhang, X. Hepatitis B virus X protein protects hepatoma and hepatic cells from complement-dependent cytotoxicity by up-regulation of CD46. FEBS Lett. 2013, 587, 645–651. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.-Y.; Lieber, A. Adenovirus Binding to Blood Factors Results in Liver Cell Infection and Hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Christiansen, D.; Devaux, P.; Réveil, B.; Evlashev, A.; Horvat, B.; Lamy, J.; Rabourdin-Combe, C.; Cohen, J.H.M.; Gerlier, D. Octamerization Enables Soluble CD46 Receptor To Neutralize Measles Virus In Vitro and In Vivo. J. Virol. 2000, 74, 4672–4678. [Google Scholar] [CrossRef]
- Varghese, P.M.; Murugaiah, V.; Beirag, N.; Temperton, N.; Khan, H.A.; Alrokayan, S.H.; Al-Ahdal, M.N.; Nal, B.; Al-Mohanna, F.A.; Sim, R.B.; et al. C4b Binding Protein Acts as an Innate Immune Effector Against Influenza A Virus. Front. Immunol. 2021, 11, 585361. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.; Murray, J.; Willard, H.; Nolan, K.; Reid, K.; Blake, D.J.; Lindsay, S.; Bhattacharya, S.; Wright, A.; Davies, K. Genetic and physical mapping around the properdin P gene. Genomics 1991, 11, 991–996. [Google Scholar] [CrossRef]
- Smith, C.; Pangburn, M.; Vogel, C.; Müller-Eberhard, H. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. J. Biol. Chem. 1984, 259, 4582–4588. [Google Scholar] [CrossRef]
- Pangburn, M. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J. Immunol. 1989, 142, 202–207. [Google Scholar]
- De Paula, P.F.; Barbosa, J.; Junior, P.; Ferriani, V.P.L.; Latorre, M.; Nudelman, V.; Isaac, L. Ontogeny of complement regulatory proteins–concentrations of factor h, factor I, c4b-binding protein, properdin and vitronectin in healthy children of different ages and in adults. Scand. J. Immunol. 2003, 58, 572–577. [Google Scholar] [CrossRef]
- Smith, K.F.; Nolan, K.F.; Reid, K.B.; Perkins, S.J. Neutron and X-ray scattering studies on the human complement protein properdin provide an analysis of the thrombospondin repeat. Biochemistry 1991, 30, 8000–8008. [Google Scholar] [CrossRef]
- Perdikoulis, M.V.; Kishore, U.; Reid, K.B. Expression and characterisation of the thrombospondin type I repeats of human properdin. Biochim. Et Biophys. Acta (Bba)-Protein Struct. Mol. Enzymol. 2001, 1548, 265–277. [Google Scholar] [CrossRef]
- Hourcade, D.E. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J. Biol. Chem. 2006, 281, 2128–2132. [Google Scholar] [CrossRef]
- Stover, C.M.; Luckett, J.C.; Echtenacher, B.; Dupont, A.; Figgitt, S.E.; Brown, J.; Männel, D.N.; Schwaeble, W.J. Properdin plays a protective role in polymicrobial septic peritonitis. J. Immunol. 2008, 180, 3313–3318. [Google Scholar] [CrossRef]
- Wirthmueller, U.; Dewald, B.; Thelen, M.; Schäfer, M.; Stover, C.; Whaley, K.; North, J.; Eggleton, P.; Reid, K.; Schwaeble, W.J. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 1997, 158, 4444–4451. [Google Scholar]
- Schwaeble, W.; Dippold, W.G.; Schäfer, M.; Pohla, H.; Jonas, D.; Luttig, B.; Weihe, E.; Huemer, H.P.; Dierich, M.P.; Reid, K. Properdin, a positive regulator of complement activation, is expressed in human T cell lines and peripheral blood T cells. J. Immunol. 1993, 151, 2521–2528. [Google Scholar]
- Schwaeble, W.; Huemer, H.P.; MÖST, J.; Dierich, M.P.; STRÖBEL, M.; Claus, C.; Reid, K.B.; ZIEGLER-HEITBROCK, H.L. Expression of properdin in human monocytes. Eur. J. Biochem. 1994, 219, 759–764. [Google Scholar] [CrossRef]
- Reis, E.; Barbuto, J.; Isaac, L. Human monocyte-derived dendritic cells are a source of several complement proteins. Inflamm. Res. 2006, 55, 179–184. [Google Scholar] [CrossRef]
- Linton, S.; Morgan, B. Properdin deficiency and meningococcal disease—identifying those most at risk. Clin. Exp. Immunol. 1999, 118, 189. [Google Scholar] [CrossRef]
- Kimura, Y.; Miwa, T.; Zhou, L.; Song, W.-C. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. BloodJ. Am. Soc. Hematol. 2008, 111, 732–740. [Google Scholar] [CrossRef]
- Kouser, L.; Abdul-Aziz, M.; Tsolaki, A.G.; Singhal, D.; Schwaeble, W.J.; Urban, B.C.; Khan, H.A.; Sim, R.B.; Kishore, U. A recombinant two-module form of human properdin is an inhibitor of the complement alternative pathway. Mol. Immunol. 2016, 73, 76–87. [Google Scholar] [CrossRef]
- Kemper, C.; Mitchell, L.M.; Zhang, L.; Hourcade, D.E. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9023–9028. [Google Scholar] [CrossRef]
- Saggu, G.; Cortes, C.; Emch, H.N.; Ramirez, G.; Worth, R.G.; Ferreira, V.P. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3 (H2O). J. Immunol. 2013, 190, 6457–6467. [Google Scholar] [CrossRef]
- Kemper, C.; Atkinson, J.P.; Hourcade, D.E. Properdin: Emerging roles of a pattern-recognition molecule. Annu. Rev. Immunol. 2009, 28, 131–155. [Google Scholar] [CrossRef]
- Jeon, H.; Yoo, S.-M.; Choi, H.S.; Mun, J.Y.; Kang, H.-G.; Lee, J.; Park, J.; Gao, S.-J.; Lee, M.-S. Extracellular vesicles from KSHV-infected endothelial cells activate the complement system. Oncotarget 2017, 8, 99841–99860. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Mackow, E.R. Endothelial Cells Elicit Immune-Enhancing Responses to Dengue Virus Infection. J. Virol. 2012, 86, 6408–6415. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, H.; Schneider, R.; Janatova, J.; Dierich, M.P. Human complement proteins C3b, C4b, factor H and properdin react with specific sites in gpl20 and gp4l, the envelope proteins of HIV1. Immunobiology 1995, 193, 98–113. [Google Scholar] [CrossRef]
- Narni-Mancinelli, E.; Gauthier, L.; Baratin, M.; Guia, S.; Fenis, A.; Deghmane, A.-E.; Rossi, B.; Fourquet, P.; Escalière, B.; Kerdiles, Y.M. Complement factor P is a ligand for the natural killer cell–activating receptor NKp46. Sci. Immunol. 2017, 2, eaam9628. [Google Scholar] [CrossRef] [PubMed]
- Beli, E. Natural Killer Cell Responses to Influenza Virus Infection in Aged Mice. Ph.D. Thesis, Michigan State University, East Lansing, MI, USA, 2012. [Google Scholar]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The natural cytotoxicity receptors in health and disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef]
- Guzzo, C.; Fox, J.; Lin, Y.; Miao, H.; Cimbro, R.; Volkman, B.F.; Fauci, A.S.; Lusso, P. The CD8-derived chemokine XCL1/lymphotactin is a conformation-dependent, broad-spectrum inhibitor of HIV-1. PLoS Pathog. 2013, 9, e1003852. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Wang, J.; Chen, M. Metabolic reprogramming in CD8+ T cells during acute viral infections. Front. Immunol. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Decker, P. Neutrophils and interferon-α-producing cells: Who produces interferon in lupus? Arthritis Res. Ther. 2011, 13, 1–3. [Google Scholar] [CrossRef]
- Kopp, A.; Hebecker, M.; Svobodova, E.; Jozsi, M. Factor h: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef]
- Fearon, D.T. Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc. Natl. Acad. Sci. USA 1978, 75, 1971–1975. [Google Scholar] [CrossRef]
- Rodriguez de Cordoba, S.; Esparza-Gordillo, J.; Goicoechea de Jorge, E.; Lopez-Trascasa, M.; Sanchez-Corral, P. The human complement factor H: Functional roles, genetic variations and disease associations. Mol. Immunol. 2004, 41, 355–367. [Google Scholar] [CrossRef]
- Pangburn, M.K.; Pangburn, K.L.; Koistinen, V.; Meri, S.; Sharma, A.K. Molecular mechanisms of target recognition in an innate immune system: Interactions among factor H, C3b, and target in the alternative pathway of human complement. J. Immunol. 2000, 164, 4742–4751. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.L.; Kaufman, R.M.; Blackmore, T.K.; Kwong, J.; Lublin, D.M. Identification of complement regulatory domains in human factor H. J. Immunol. 1995, 155, 348–356. [Google Scholar] [PubMed]
- McRae, J.L.; Duthy, T.G.; Griggs, K.M.; Ormsby, R.J.; Cowan, P.J.; Cromer, B.A.; McKinstry, W.J.; Parker, M.W.; Murphy, B.F.; Gordon, D.L. Human factor H-related protein 5 has cofactor activity, inhibits C3 convertase activity, binds heparin and C-reactive protein, and associates with lipoprotein. J. Immunol. 2005, 174, 6250–6256. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Jokiranta, T.S.; Hellwage, J.; Koistinen, V.; Meri, S. The factor H protein family. Immunopharmacology 1999, 42, 53–60. [Google Scholar] [CrossRef]
- DeCordova, S.; Abdelgany, A.; Murugaiah, V.; Pathan, A.A.; Nayak, A.; Walker, T.; Shastri, A.; Alrokayan, S.H.; Khan, H.A.; Singh, S.K.; et al. Secretion of functionally active complement factor H related protein 5 (FHR5) by primary tumour cells derived from Glioblastoma Multiforme patients. Immunobiology 2019, 224, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, R.L.; Griffin, D.E.; Winkelstein, J.A. Host modification of Sindbis virus sialic acid content influences alternative complement pathway activation and virus clearance. J. Immunol. 1981, 127, 1740–1743. [Google Scholar]
- Pinter, C.; Siccardi, A.G.; Longhi, R.; Clivio, A. Direct interaction of complement factor H with the C1 domain of HIV type 1 glycoprotein 120. AIDS Res. Hum. Retrovir. 1995, 11, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Lambris, J.D.; Ricklin, D.; Geisbrecht, B.V. Complement evasion by human pathogens. Nat. Rev. Microbiol. 2008, 6, 132–142. [Google Scholar] [CrossRef]
- Brinton, M.A. Replication cycle and molecular biology of the West Nile virus. Viruses 2013, 6, 13–53. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile Virus: Biology, transmission, and human infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Murugaiah, V.; Varghese, P.M.; Saleh, S.M.; Tsolaki, A.G.; Alrokayan, S.H.; Khan, H.A.; Collison, K.S.; Sim, R.B.; Nal, B.; Al-Mohanna, F.A.; et al. Complement-Independent Modulation of Influenza A Virus Infection by Factor H. Front. Immunol. 2020, 11, 355. [Google Scholar] [CrossRef]
- Kuhlman, M.; Joiner, K.; Ezekowitz, R.A. The human mannose-binding protein functions as an opsonin. J. Exp. Med. 1989, 169, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Chang, W.-C.; Takahashi, M.; Pavlov, V.; Ishida, Y.; La Bonte, L.; Shi, L.; Fujita, T.; Stahl, G.L.; Van Cott, E.M. Mannose-binding lectin and its associated proteases (MASPs) mediate coagulation and its deficiency is a risk factor in developing complications from infection, including disseminated intravascular coagulation. Immunobiology 2011, 216, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ip, W.E.; Michelow, I.C.; Ezekowitz, R.A.B. The mannose-binding lectin: A prototypic pattern recognition molecule. Curr. Opin. Immunol. 2006, 18, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Murugaiah, V.; Tsolaki, A.G.; Kishore, U. Collectins: Innate Immune Pattern Recognition Molecules. Adv. Exp. Med. Biol. 2020, 1204, 75–127. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.P.; Tarr, A.W. Human lectins and their roles in viral infections. Molecules 2015, 20, 2229–2271. [Google Scholar] [CrossRef]
- Turner, M.W.; Hamvas, R.M. Mannose-binding lectin: Structure, function, genetics and disease associations. Rev. Immunogenet. 2000, 2, 305–322. [Google Scholar]
- Hansen, S.; Thiel, S.; Willis, A.; Holmskov, U.; Jensenius, J.C. Purification and characterization of two mannan-binding lectins from mouse serum. J. Immunol. 2000, 164, 2610–2618. [Google Scholar] [CrossRef]
- Sastry, K.; Zahedi, K.; Lelias, J.M.; Whitehead, A.S.; Ezekowitz, R.A. Molecular characterization of the mouse mannose-binding proteins. The mannose-binding protein A but not C is an acute phase reactant. J. Immunol. 1991, 147, 692–697. [Google Scholar]
- Ezekowitz, R.A.; Day, L.E.; Herman, G.A. A human mannose-binding protein is an acute-phase reactant that shares sequence homology with other vertebrate lectins. J. Exp Med 1988, 167, 1034–1046. [Google Scholar] [CrossRef]
- Singh, K.K.; Nathamu, S.; Adame, A.; Alire, T.U.; Dumaop, W.; Gouaux, B.; Moore, D.J.; Masliah, E. Expression of mannose binding lectin in HIV-1-infected brain: Implications for HIV-related neuronal damage and neuroAIDS. Neurobehav. HIV Med. 2011, 3, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Mogues, T.; Ota, T.; Tauber, A.I.; Sastry, K.N. Characterization of two mannose-binding protein cDNAs from rhesus monkey (Macaca mulatta): Structure and evolutionary implications. Glycobiology 1996, 6, 543–550. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Drickamer, K.; Dordal, M.S.; Reynolds, L. Mannose-binding proteins isolated from rat liver contain carbohydrate-recognition domains linked to collagenous tails. Complete primary structures and homology with pulmonary surfactant apoprotein. J. Biol. Chem. 1986, 261, 6878–6887. [Google Scholar] [CrossRef]
- Shi, L.; Takahashi, K.; Dundee, J.; Shahroor-Karni, S.; Thiel, S.; Jensenius, J.C.; Gad, F.; Hamblin, M.R.; Sastry, K.N.; Ezekowitz, R.A. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J. Exp. Med. 2004, 199, 1379–1390. [Google Scholar] [CrossRef]
- Reading, P.C.; Hartley, C.A.; Ezekowitz, R.A.; Anders, E.M. A serum mannose-binding lectin mediates complement-dependent lysis of influenza virus-infected cells. Biochem. Biophys. Res. Commun. 1995, 217, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Reading, P.C.; Morey, L.S.; Crouch, E.C.; Anders, E.M. Collectin-mediated antiviral host defense of the lung: Evidence from influenza virus infection of mice. J. Virol. 1997, 71, 8204–8212. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, K.L.; Sastry, K.; White, M.R.; Anders, E.M.; Super, M.; Ezekowitz, R.A.; Tauber, A.I. Human mannose-binding protein functions as an opsonin for influenza A viruses. J. Clin. Investig. 1993, 91, 1414–1420. [Google Scholar] [CrossRef]
- Hartley, C.A.; Jackson, D.C.; Anders, E.M. Two distinct serum mannose-binding lectins function as beta inhibitors of influenza virus: Identification of bovine serum beta inhibitor as conglutinin. J. Virol. 1992, 66, 4358–4363. [Google Scholar] [CrossRef]
- Fidler, K.J.; Hilliard, T.N.; Bush, A.; Johnson, M.; Geddes, D.M.; Turner, M.W.; Alton, E.W.; Klein, N.J.; Davies, J.C. Mannose-binding lectin is present in the infected airway: A possible pulmonary defence mechanism. Thorax 2009, 64, 150–155. [Google Scholar] [CrossRef]
- Kase, T.; Suzuki, Y.; Kawai, T.; Sakamoto, T.; Ohtani, K.; Eda, S.; Maeda, A.; Okuno, Y.; Kurimura, T.; Wakamiya, N. Human mannan-binding lectin inhibits the infection of influenza A virus without complement. Immunology 1999, 97, 385–392. [Google Scholar] [CrossRef]
- Anders, E.M.; Hartley, C.A.; Reading, P.C.; Ezekowitz, R.A. Complement-dependent neutralization of influenza virus by a serum mannose-binding lectin. J. Gen. Virol. 1994, 75 Pt 3, 615–622. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Murugaiah, V.; Bashir, H.A.; Pathan, A.A.; Abozaid, S.M.; Makarov, E.; Nal-Rogier, B.; Kishore, U.; Al-Ahdal, M.N. Full-length human surfactant protein A inhibits influenza A virus infection of A549 lung epithelial cells: A recombinant form containing neck and lectin domains promotes infectivity. Immunobiology 2019, 224, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahdal, M.N.; Murugaiah, V.; Varghese, P.M.; Abozaid, S.M.; Saba, I.; Al-Qahtani, A.A.; Pathan, A.A.; Kouser, L.; Nal, B.; Kishore, U. Entry Inhibition and Modulation of Pro-Inflammatory Immune Response Against Influenza A Virus by a Recombinant Truncated Surfactant Protein D. Front. Immunol. 2018, 9, 1586. [Google Scholar] [CrossRef] [PubMed]
- Crouch, E.; Hartshorn, K.; Horlacher, T.; McDonald, B.; Smith, K.; Cafarella, T.; Seaton, B.; Seeberger, P.H.; Head, J. Recognition of mannosylated ligands and influenza A virus by human surfactant protein D: Contributions of an extended site and residue 343. Biochemistry 2009, 48, 3335–3345. [Google Scholar] [CrossRef]
- Hartshorn, K.L.; Webby, R.; White, M.R.; Tecle, T.; Pan, C.; Boucher, S.; Moreland, R.J.; Crouch, E.C.; Scheule, R.K. Role of viral hemagglutinin glycosylation in anti-influenza activities of recombinant surfactant protein D. Respir. Res. 2008, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, K.L.; Crouch, E.C.; White, M.R.; Eggleton, P.; Tauber, A.I.; Chang, D.; Sastry, K. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Investig. 1994, 94, 311–319. [Google Scholar] [CrossRef]
- Tokunaga, H.; Ushirogawa, H.; Ohuchi, M. The pandemic (H1N1) 2009 influenza virus is resistant to mannose-binding lectin. Virol. J. 2011, 8, 50. [Google Scholar] [CrossRef]
- Job, E.R.; Deng, Y.M.; Tate, M.D.; Bottazzi, B.; Crouch, E.C.; Dean, M.M.; Mantovani, A.; Brooks, A.G.; Reading, P.C. Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J. Immunol. 2010, 185, 4284–4291. [Google Scholar] [CrossRef]
- Chang, W.C.; White, M.R.; Moyo, P.; McClear, S.; Thiel, S.; Hartshorn, K.L.; Takahashi, K. Lack of the pattern recognition molecule mannose-binding lectin increases susceptibility to influenza A virus infection. Bmc Immunol. 2010, 11, 64. [Google Scholar] [CrossRef]
- Ling, M.T.; Tu, W.; Han, Y.; Mao, H.; Chong, W.P.; Guan, J.; Liu, M.; Lam, K.T.; Law, H.K.; Peiris, J.S.; et al. Mannose-binding lectin contributes to deleterious inflammatory response in pandemic H1N1 and avian H9N2 infection. J. Infect. Dis. 2012, 205, 44–53. [Google Scholar] [CrossRef]
- Saifuddin, M.; Hart, M.L.; Gewurz, H.; Zhang, Y.; Spear, G.T. Interaction of mannose-binding lectin with primary isolates of human immunodeficiency virus type 1. J. Gen. Virol. 2000, 81, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Mayer, C.; Brown, A.; Harouse, J.; Luciw, P.A.; Mayer, A.J. Selection for neutralization resistance of the simian/human immunodeficiency virus SHIVSF33A variant in vivo by virtue of sequence changes in the extracellular envelope glycoprotein that modify N-linked glycosylation. J. Virol. 1999, 73, 5294–5300. [Google Scholar] [CrossRef] [PubMed]
- Reitter, J.N.; Means, R.E.; Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998, 4, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Rudensey, L.M.; Overbaugh, J. Specific N-linked and O-linked glycosylation modifications in the envelope V1 domain of simian immunodeficiency virus variants that evolve in the host alter recognition by neutralizing antibodies. J. Virol. 1997, 71, 7719–7727. [Google Scholar] [CrossRef] [PubMed]
- Jack, D.L.; Lee, M.E.; Turner, M.W.; Klein, N.J.; Read, R.C. Mannose-binding lectin enhances phagocytosis and killing of Neisseria meningitidis by human macrophages. J. Leukoc. Biol. 2005, 77, 328–336. [Google Scholar] [CrossRef]
- Ying, H.; Ji, X.; Hart, M.L.; Gupta, K.; Saifuddin, M.; Zariffard, M.R.; Spear, G.T. Interaction of mannose-binding lectin with HIV type 1 is sufficient for virus opsonization but not neutralization. AIDS Res. Hum. Retrovir. 2004, 20, 327–335. [Google Scholar] [CrossRef]
- Senaldi, G.; Davies, E.T.; Mahalingam, M.; Lu, J.; Pozniak, A.; Peakman, M.; Reid, K.B.; Vergani, D. Circulating levels of mannose binding protein in human immunodeficiency virus infection. J. Infect. 1995, 31, 145–148. [Google Scholar] [CrossRef]
- Ballegaard, V.; Haugaard, A.K.; Garred, P.; Nielsen, S.D.; Munthe-Fog, L. The lectin pathway of complement: Advantage or disadvantage in HIV pathogenesis? Clin. Immunol. 2014, 154, 13–25. [Google Scholar] [CrossRef]
- Takahashi, K.; Ezekowitz, R.A. The role of the mannose-binding lectin in innate immunity. Clin. Infect. Dis. 2005, 41 (Suppl. 7), S440–S444. [Google Scholar] [CrossRef]
- Garred, P.; Madsen, H.O.; Balslev, U.; Hofmann, B.; Pedersen, C.; Gerstoft, J.; Svejgaard, A. Susceptibility to HIV infection and progression of AIDS in relation to variant alleles of mannose-binding lectin. Lancet 1997, 349, 236–240. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, K.; Pfefferle, S.; Bertram, S.; Glowacka, I.; Drosten, C.; Pohlmann, S.; Simmons, G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010, 84, 8753–8764. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.; Chan, K.H.; Law, H.K.; Tso, G.H.; Kong, E.K.; Wong, W.H.; To, Y.F.; Yung, R.W.; Chow, E.Y.; Au, K.L.; et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 2005, 191, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Avirutnan, P.; Hauhart, R.E.; Marovich, M.A.; Garred, P.; Atkinson, J.P.; Diamond, M.S. Complement-mediated neutralization of dengue virus requires mannose-binding lectin. mBio 2011, 2. [Google Scholar] [CrossRef]
- Fuchs, A.; Lin, T.Y.; Beasley, D.W.; Stover, C.M.; Schwaeble, W.J.; Pierson, T.C.; Diamond, M.S. Direct complement restriction of flavivirus infection requires glycan recognition by mannose-binding lectin. Cell Host Microbe 2010, 8, 186–195. [Google Scholar] [CrossRef]
- Brown, K.S.; Keogh, M.J.; Owsianka, A.M.; Adair, R.; Patel, A.H.; Arnold, J.N.; Ball, J.K.; Sim, R.B.; Tarr, A.W.; Hickling, T.P. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell 2010, 1, 664–674. [Google Scholar] [CrossRef]
- Teodorof, C.; Divakar, S.; Soontornniyomkij, B.; Achim, C.L.; Kaul, M.; Singh, K.K. Intracellular mannose binding lectin mediates subcellular trafficking of HIV-1 gp120 in neurons. Neurobiol. Dis. 2014, 69, 54–64. [Google Scholar] [CrossRef]
- Bachis, A.; Aden, S.A.; Nosheny, R.L.; Andrews, P.M.; Mocchetti, I. Axonal transport of human immunodeficiency virus type 1 envelope protein glycoprotein 120 is found in association with neuronal apoptosis. J. Neurosci. 2006, 26, 6771–6780. [Google Scholar] [CrossRef]
- Hakozaki, Y.; Yoshiba, M.; Sekiyama, K.; Seike, E.; Iwamoto, J.; Mitani, K.; Mine, M.; Morizane, T.; Ohtani, K.; Suzuki, Y.; et al. Mannose-binding lectin and the prognosis of fulminant hepatic failure caused by HBV infection. Liver 2002, 22, 29–34. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsutsumi, A.; Wakamiya, N.; Ohtani, K.; Suzuki, Y.; Watanabe, Y.; Nakayama, N.; Koike, T. Mannose-binding lectin polymorphisms in patients with hepatitis C virus infection. Scand. J. Gastroenterol. 2000, 35, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Lau, C.S.; Lau, Y.L.; Wong, W.M.; Cheng, C.C.; Lai, C.L. Mannose binding lectin gene mutations are associated with progression of liver disease in chronic hepatitis B infection. Hepatology 1999, 29, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Hijikata, M.; Matsushita, M.; Ohta, Y.; Mishiro, S. Association of mannose-binding lectin gene haplotype LXPA and LYPB with interferon-resistant hepatitis C virus infection in Japanese patients. J. Hepatol. 1998, 29, 695–700. [Google Scholar] [CrossRef]
- Thomas, H.C.; Foster, G.R.; Sumiya, M.; McIntosh, D.; Jack, D.L.; Turner, M.W.; Summerfield, J.A. Mutation of gene of mannose-binding protein associated with chronic hepatitis B viral infection. Lancet 1996, 348, 1417–1419. [Google Scholar] [CrossRef]
- Hohler, T.; Wunschel, M.; Gerken, G.; Schneider, P.M.; Meyer zum Buschenfelde, K.H.; Rittner, C. No association between mannose-binding lectin alleles and susceptibility to chronic hepatitis B virus infection in German patients. Exp. Clin. Immunogenet. 1998, 15, 130–133. [Google Scholar] [CrossRef]
- Meyer, K.; Basu, A.; Przysiecki, C.T.; Lagging, L.M.; Di Bisceglie, A.M.; Conley, A.J.; Ray, R. Complement-mediated enhancement of antibody function for neutralization of pseudotype virus containing hepatitis C virus E2 chimeric glycoprotein. J. Virol. 2002, 76, 2150–2158. [Google Scholar] [CrossRef]
- Chong, W.P.; To, Y.F.; Ip, W.K.; Yuen, M.F.; Poon, T.P.; Wong, W.H.; Lai, C.L.; Lau, Y.L. Mannose-binding lectin in chronic hepatitis B virus infection. Hepatology 2005, 42, 1037–1045. [Google Scholar] [CrossRef]
- Gadjeva, M.; Paludan, S.R.; Thiel, S.; Slavov, V.; Ruseva, M.; Eriksson, K.; Lowhagen, G.B.; Shi, L.; Takahashi, K.; Ezekowitz, A.; et al. Mannan-binding lectin modulates the response to HSV-2 infection. Clin. Exp. Immunol. 2004, 138, 304–311. [Google Scholar] [CrossRef]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and adaptive immune responses to herpes simplex virus. Viruses 2009, 1, 979–1002. [Google Scholar] [CrossRef]
- Biron, C.A.; Brossay, L. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 2001, 13, 458–464. [Google Scholar] [CrossRef]
- Cerwenka, A.; Lanier, L.L. Ligands for natural killer cell receptors: Redundancy or specificity. Immunol. Rev. 2001, 181, 158–169. [Google Scholar] [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Seppanen, M.; Lokki, M.L.; Lappalainen, M.; Hiltunen-Back, E.; Rovio, A.T.; Kares, S.; Hurme, M.; Aittoniemi, J. Mannose-binding lectin 2 gene polymorphism in recurrent herpes simplex virus 2 infection. Hum. Immunol. 2009, 70, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Gramberg, T.; Simmons, G.; Moller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D., Jr.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [PubMed]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Escudero-Perez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef]
- Michelow, I.C.; Lear, C.; Scully, C.; Prugar, L.I.; Longley, C.B.; Yantosca, L.M.; Ji, X.; Karpel, M.; Brudner, M.; Takahashi, K.; et al. High-dose mannose-binding lectin therapy for Ebola virus infection. J. Infect Dis. 2011, 203, 175–179. [Google Scholar] [CrossRef]
- Fuchs, A.; Pinto, A.K.; Schwaeble, W.J.; Diamond, M.S. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 2011, 412, 101–109. [Google Scholar] [CrossRef]
- Hummelshoj, T.; Thielens, N.M.; Madsen, H.O.; Arlaud, G.J.; Sim, R.B.; Garred, P. Molecular organization of human Ficolin-2. Mol. Immunol. 2007, 44, 401–411. [Google Scholar] [CrossRef]
- Matsushita, M.; Endo, Y.; Taira, S.; Sato, Y.; Fujita, T.; Ichikawa, N.; Nakata, M.; Mizuochi, T. A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. J. Biol. Chem. 1996, 271, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Drickamer, K. Trimeric structure of a C-type mannose-binding protein. Structure 1994, 2, 1227–1240. [Google Scholar] [CrossRef]
- Sheriff, S.; Chang, C.Y.; Ezekowitz, R.A. Human mannose-binding protein carbohydrate recognition domain trimerizes through a triple alpha-helical coiled-coil. Nat. Struct. Biol. 1994, 1, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Erickson, H.P. The disulfide bonding pattern in ficolin multimers. J. Biol. Chem. 2004, 279, 6534–6539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Ali, M.A. Ficolins: Structure, function and associated diseases. Adv. Exp. Med. Biol. 2008, 632, 105–115. [Google Scholar] [PubMed]
- Akaiwa, M.; Yae, Y.; Sugimoto, R.; Suzuki, S.O.; Iwaki, T.; Izuhara, K.; Hamasaki, N. Hakata antigen, a new member of the ficolin/opsonin p35 family, is a novel human lectin secreted into bronchus/alveolus and bile. J. Histochem. Cytochem. 1999, 47, 777–786. [Google Scholar] [CrossRef]
- Ren, Y.; Ding, Q.; Zhang, X. Ficolins and infectious diseases. Virol. Sin. 2014, 29, 25–32. [Google Scholar] [CrossRef]
- Matsushita, M. Ficolins in complement activation. Mol. Immunol. 2013, 55, 22–26. [Google Scholar] [CrossRef]
- Garlatti, V.; Belloy, N.; Martin, L.; Lacroix, M.; Matsushita, M.; Endo, Y.; Fujita, T.; Fontecilla-Camps, J.C.; Arlaud, G.J.; Thielens, N.M.; et al. Structural insights into the innate immune recognition specificities of L- and H-ficolins. EMBO J. 2007, 26, 623–633. [Google Scholar] [CrossRef]
- Liu, Y.; Endo, Y.; Iwaki, D.; Nakata, M.; Matsushita, M.; Wada, I.; Inoue, K.; Munakata, M.; Fujita, T. Human M-ficolin is a secretory protein that activates the lectin complement pathway. J. Immunol. 2005, 175, 3150–3156. [Google Scholar] [CrossRef]
- Swierzko, A.; Lukasiewicz, J.; Cedzynski, M.; Maciejewska, A.; Jachymek, W.; Niedziela, T.; Matsushita, M.; Lugowski, C. New functional ligands for ficolin-3 among lipopolysaccharides of Hafnia alvei. Glycobiology 2012, 22, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Honore, C.; Rorvig, S.; Hummelshoj, T.; Skjoedt, M.O.; Borregaard, N.; Garred, P. Tethering of Ficolin-1 to cell surfaces through recognition of sialic acid by the fibrinogen-like domain. J. Leukoc. Biol. 2010, 88, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.G.; Cho, M.Y.; Zhao, M.; Park, J.W.; Matsushita, M.; Fujita, T.; Lee, B.L. Human mannose-binding lectin and L-ficolin function as specific pattern recognition proteins in the lectin activation pathway of complement. J. Biol. Chem. 2004, 279, 25307–25312. [Google Scholar] [CrossRef] [PubMed]
- Lynch, N.J.; Roscher, S.; Hartung, T.; Morath, S.; Matsushita, M.; Maennel, D.N.; Kuraya, M.; Fujita, T.; Schwaeble, W.J. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-positive bacteria, and activates the lectin pathway of complement. J. Immunol. 2004, 172, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, R.; Yae, Y.; Akaiwa, M.; Kitajima, S.; Shibata, Y.; Sato, H.; Hirata, J.; Okochi, K.; Izuhara, K.; Hamasaki, N. Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. J. Biol. Chem. 1998, 273, 20721–20727. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Sun, X.; Wang, Y.; Wang, Q.; Wu, Y.; Pan, Q.; Fang, C.; Zhang, X.L. Ficolin-2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLoS ONE 2013, 8, e73859. [Google Scholar] [CrossRef]
- Pan, Q.; Chen, H.; Wang, F.; Jeza, V.T.; Hou, W.; Zhao, Y.; Xiang, T.; Zhu, Y.; Endo, Y.; Fujita, T.; et al. L-ficolin binds to the glycoproteins hemagglutinin and neuraminidase and inhibits influenza A virus infection both in vitro and in vivo. J. Innate Immun. 2012, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Keirstead, N.D.; Lee, C.; Yoo, D.; Brooks, A.S.; Hayes, M.A. Porcine plasma ficolin binds and reduces infectivity of porcine reproductive and respiratory syndrome virus (PRRSV) in vitro. Antivir. Res. 2008, 77, 28–38. [Google Scholar] [CrossRef]
- Verma, A.; White, M.; Vathipadiekal, V.; Tripathi, S.; Mbianda, J.; Ieong, M.; Qi, L.; Taubenberger, J.K.; Takahashi, K.; Jensenius, J.C.; et al. Human H-ficolin inhibits replication of seasonal and pandemic influenza A viruses. J. Immunol. 2012, 189, 2478–2487. [Google Scholar] [CrossRef]
- Liu, J.; Ali, M.A.; Shi, Y.; Zhao, Y.; Luo, F.; Yu, J.; Xiang, T.; Tang, J.; Li, D.; Hu, Q.; et al. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell Mol. Immunol. 2009, 6, 235–244. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, Y.; Zhang, X.; Zhao, P.; Tao, W.; Zhong, J.; Li, Q.; Zhang, X.L. Ficolin-2 inhibits hepatitis C virus infection, whereas apolipoprotein E3 mediates viral immune escape. J. Immunol. 2014, 193, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.R.; Brown, R.J.; Zothner, C.; Urbanowicz, R.A.; Mason, C.P.; Krarup, A.; McClure, C.P.; Irving, W.L.; Ball, J.K.; Harris, M.; et al. Recombinant human L-ficolin directly neutralizes hepatitis C virus entry. J. Innate Immun. 2014, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Gout, E.; Moriscot, C.; Doni, A.; Dumestre-Perard, C.; Lacroix, M.; Perard, J.; Schoehn, G.; Mantovani, A.; Arlaud, G.J.; Thielens, N.M. M-ficolin interacts with the long pentraxin PTX3: A novel case of cross-talk between soluble pattern-recognition molecules. J. Immunol. 2011, 186, 5815–5822. [Google Scholar] [CrossRef] [PubMed]
- Favier, A.L.; Gout, E.; Reynard, O.; Ferraris, O.; Kleman, J.P.; Volchkov, V.; Peyrefitte, C.; Thielens, N.M. Enhancement of Ebola Virus Infection via Ficolin-1 Interaction with the Mucin Domain of GP Glycoprotein. J. Virol. 2016, 90, 5256–5269. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Virus | C1q Binding | Consequences of C1q Binding | MBL Binding | Consequences of MBL Binding | C4BP Binding | Consequences of C4BP Binding | Properdin Binding | Consequences of Properdin Binding | Factor H Binding | Consequences of Factor H Binding |
|---|---|---|---|---|---|---|---|---|---|---|
| Murine Leukaemia Virus (MuLV) | + | Complement activation Viral lysis by human serum | ? | |||||||
| Human Immunodeficiency Virus (HIV-1, 2) | + | Complement activation No virolysis by human serum Enhancement of infection of C receptor-bearing cells | + | In vitro inhibition of the virus Complement activation Enhancement of infection Virus uptake by macrophages | ? | + | + | Escape complement destruction | ||
| Human T Lymphotropic Virus (HTLV) | + | In vitro inhibition of the virus complement activation No virolysis by human serum | + | |||||||
| Herpes Simplex Virus 1 (HSV-1) | + | Neutralisation of a gC null virus by a C1–C5 dependent mechanism | ? | |||||||
| Herpes Simplex Virus 2 (HSV-2) | ? | + | Enhancement of infection in a murine model | |||||||
| Human gamma-herpesvirus 8 | + | Induces complement activation during de novo KSHV infection | ||||||||
| Epstein-Barr Virus (EBV) | + | Classical pathway activation No neutralisation of the virus Opsonisation of the virus | ? | |||||||
| Cytomegalovirus (CMV) | + | Classical pathway activation No lysis of infected cells | - | |||||||
| Influenza A Virus (IAV) | + | + | C-independent virus inactivation Complement activation (guinea pig) | + | Differentially modulates the efficacy of viral entry and replication in a strain-dependent manner | + | Differentially modulates the efficacy of viral entry and replication in a strain-dependent manner | + | Differentially modulates the efficacy of viral entry and replication in a strain-dependent manner | |
| Flavivirus | + | Viral NS1 limits complement activation by binding C4BP | + | Enhance complement activation | Enables immune evasion | |||||
| Hepatitis B virus | + | Upregulates C4BPα through transcription factor Sp1 inhibiting complement activation | ||||||||
| Adenovirus | + | Increased uptake by hepatocytes Reduced hepatic and innate toxicity after systemic application of adenoviruses vector | ||||||||
| Sindbis virus | + | Reduced alternative pathway activation | ||||||||
| SARS CoV 2 | ? | Lower C1q levels in the blood of severely ill patients Localised deposits of C1q in lungs suggesting classical pathway activation | ? | N-protein of SARS-CoV-2 has been shown to interact with (MASP2) | ? | ? | ? | The addition of factor H help mitigate damages caused by uncontrolled alternative pathway activation by viral S1/S2 protein |
| Virus Name | Viral Protein | Host Homologue | Targets | Evasion Mechanism |
|---|---|---|---|---|
| Herpesvirus Saimiri | Complement Control Protein Homologue (CCPH) | DAF, MCP | CP/LP; AP C3 convertase | Decay AccelerationCofactor Activity |
| Herpesvirus Saimiri | HVSCD59 | CD59 | C5b-8 and C5b-9 | MAC Complex Inhibition |
| Kaposi’s Sarcoma-Associated Herpesvirus | KSHV Complement Control Protein (KCP) | DAF, MCP | CP/LP and AP C3 convertase | Decay AccelerationCofactor Activity |
| Murine Gamma-herpesvirus 68 | γ-hv68 RCA Protein | DAF, MCP | CP/LP and AP C3 convertase | Exact mechanism unknown; Effect possibly mediated through C3 interaction |
| Variola Virus | Variola Virus Inhibitor Of Complement Enzymes (SPICE) | MCP | CP/LP and AP C3/C5 convertase | Decay AccelerationCofactor Activity |
| Vaccinia Virus | Vaccinia Virus Complement Control Protein (VCP) | MCP | CP/LP and AP C3/C5 convertase | Decay AccelerationCofactor Activity |
| Monkeypox Virus | Monkeypox Virus Inhibitor Of Complement Enzymes (MOPICE) | MCP | CP/LP C3 convertase | Cofactor Activity |
| Astrovirus | Coat Protein | Human Neutrophil Defensin-1 | C1q and MBL | Classical Pathway Inactivation: Disassociates C1s from C1q Lectin Pathway Inactivation: Blocks Masp-2 interaction with MBL |
| Herpes Simplex Virus | Envelope Surface Glycoprotein C | N/A | AP C3 convertase and C3b | Blocks C3b interaction with properdin and C5 Accelerates decay of alternative pathway C3 Convertase |
| Hepatitis B virus | HBx protein | N/A | CD46, CD59 and C4BP α | MAC Complex Inhibition CP/LP C3 convertase |
| Influenza virus | Matrix Protein 1 | N/A | C1q | Classical Pathway Inactivation: Inhibits C1q interaction with IgG antibodies |
| Dengue (DENV), West Nile Virus, And Yellow Fever (YFV) | Non-Structural Protein 1 (NS1) | N/A | C1s, C4, C9 | Bind Factor H/C4BP → Cofactor Activity Decreases deposition of C3b and MAC Interacts with C4 And C1s → Reduces classical C3 convertase and reduced deposition of C4b And C3b Binds Clusterin & Vitronectin → MAC Complex Inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murugaiah, V.; Varghese, P.M.; Beirag, N.; De Cordova, S.; Sim, R.B.; Kishore, U. Complement Proteins as Soluble Pattern Recognition Receptors for Pathogenic Viruses. Viruses 2021, 13, 824. https://doi.org/10.3390/v13050824
Murugaiah V, Varghese PM, Beirag N, De Cordova S, Sim RB, Kishore U. Complement Proteins as Soluble Pattern Recognition Receptors for Pathogenic Viruses. Viruses. 2021; 13(5):824. https://doi.org/10.3390/v13050824
Chicago/Turabian StyleMurugaiah, Valarmathy, Praveen M. Varghese, Nazar Beirag, Syreeta De Cordova, Robert B. Sim, and Uday Kishore. 2021. "Complement Proteins as Soluble Pattern Recognition Receptors for Pathogenic Viruses" Viruses 13, no. 5: 824. https://doi.org/10.3390/v13050824
APA StyleMurugaiah, V., Varghese, P. M., Beirag, N., De Cordova, S., Sim, R. B., & Kishore, U. (2021). Complement Proteins as Soluble Pattern Recognition Receptors for Pathogenic Viruses. Viruses, 13(5), 824. https://doi.org/10.3390/v13050824