
CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
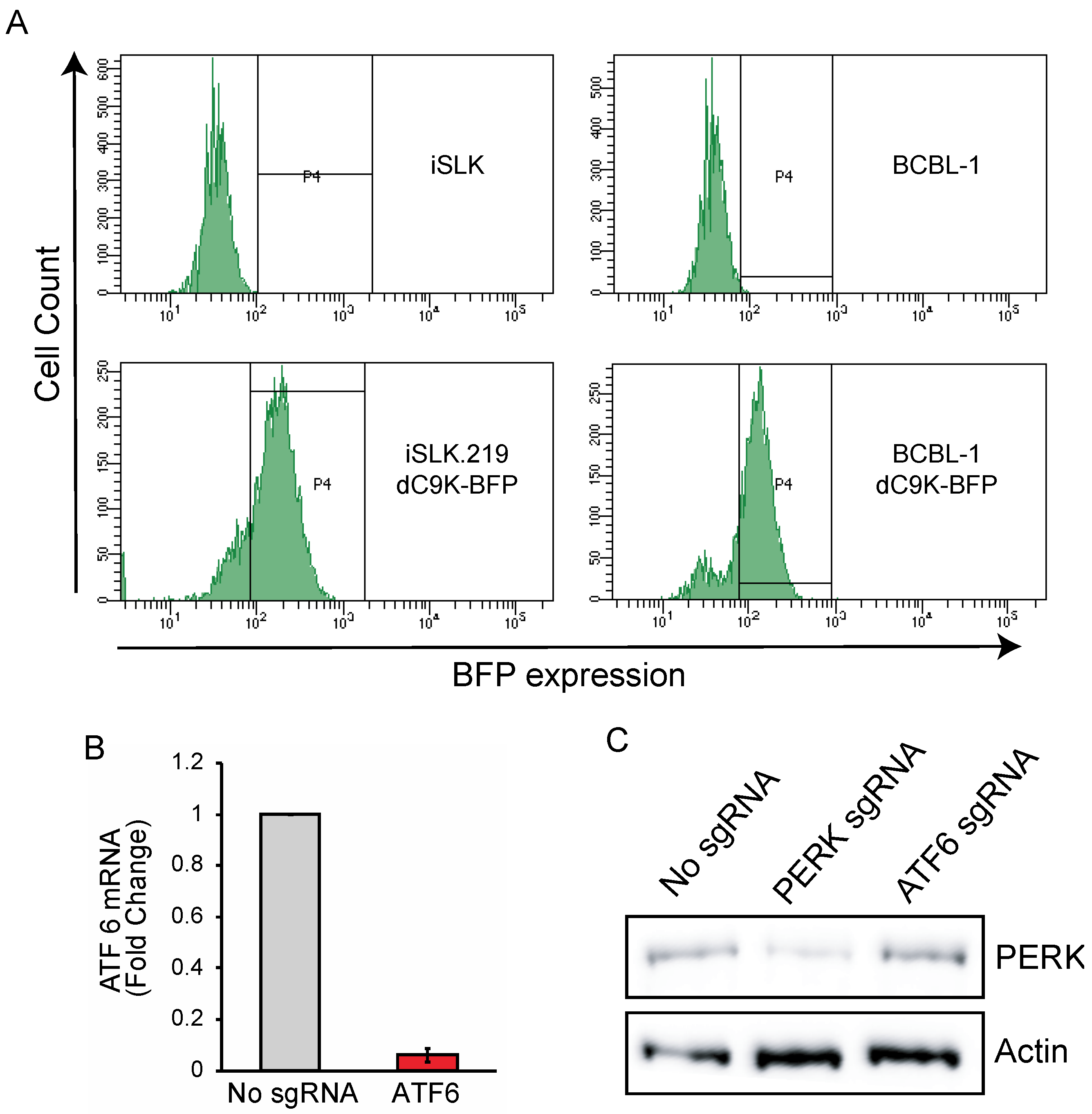
2.2. Generation of iSLK-219-dC9K and BCBL-1-dC9K
2.3. sgRNA Design and Transduction
2.4. Immunoblotting and Antibodies
2.5. Immunofluorescence
2.6. Quantitative Reverse Transcription PCR
2.7. Virus Tittering
3. Results
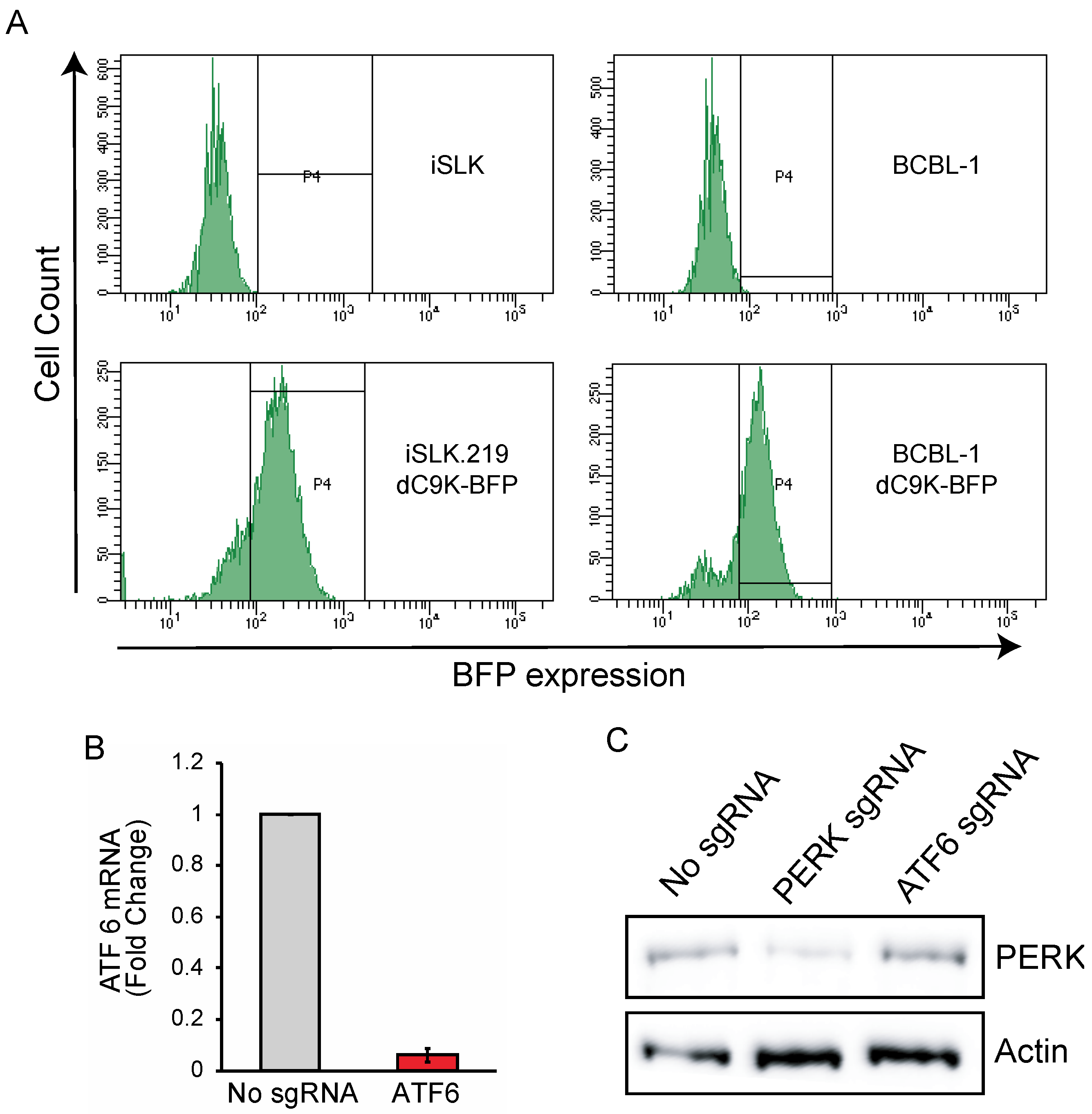
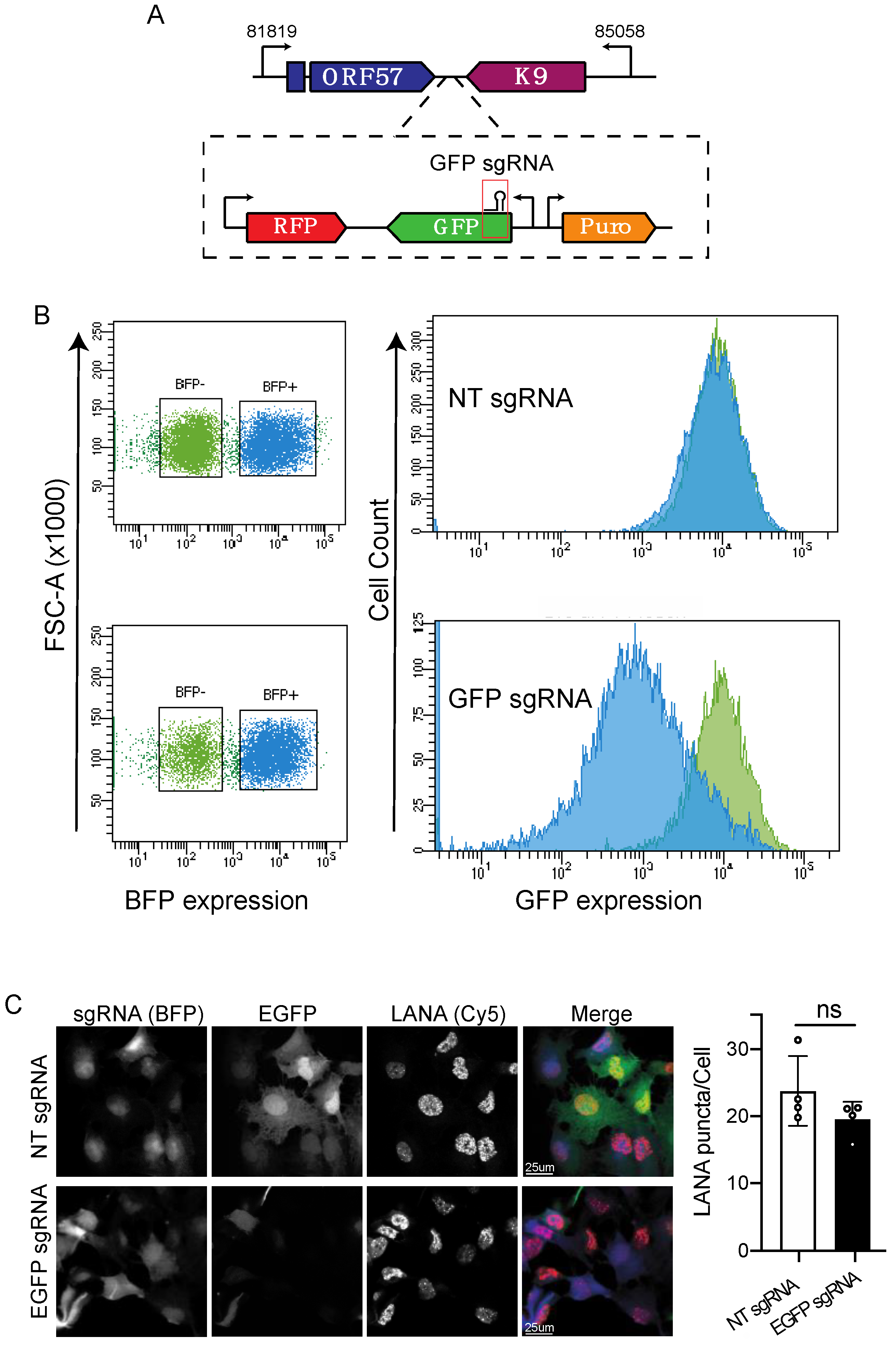
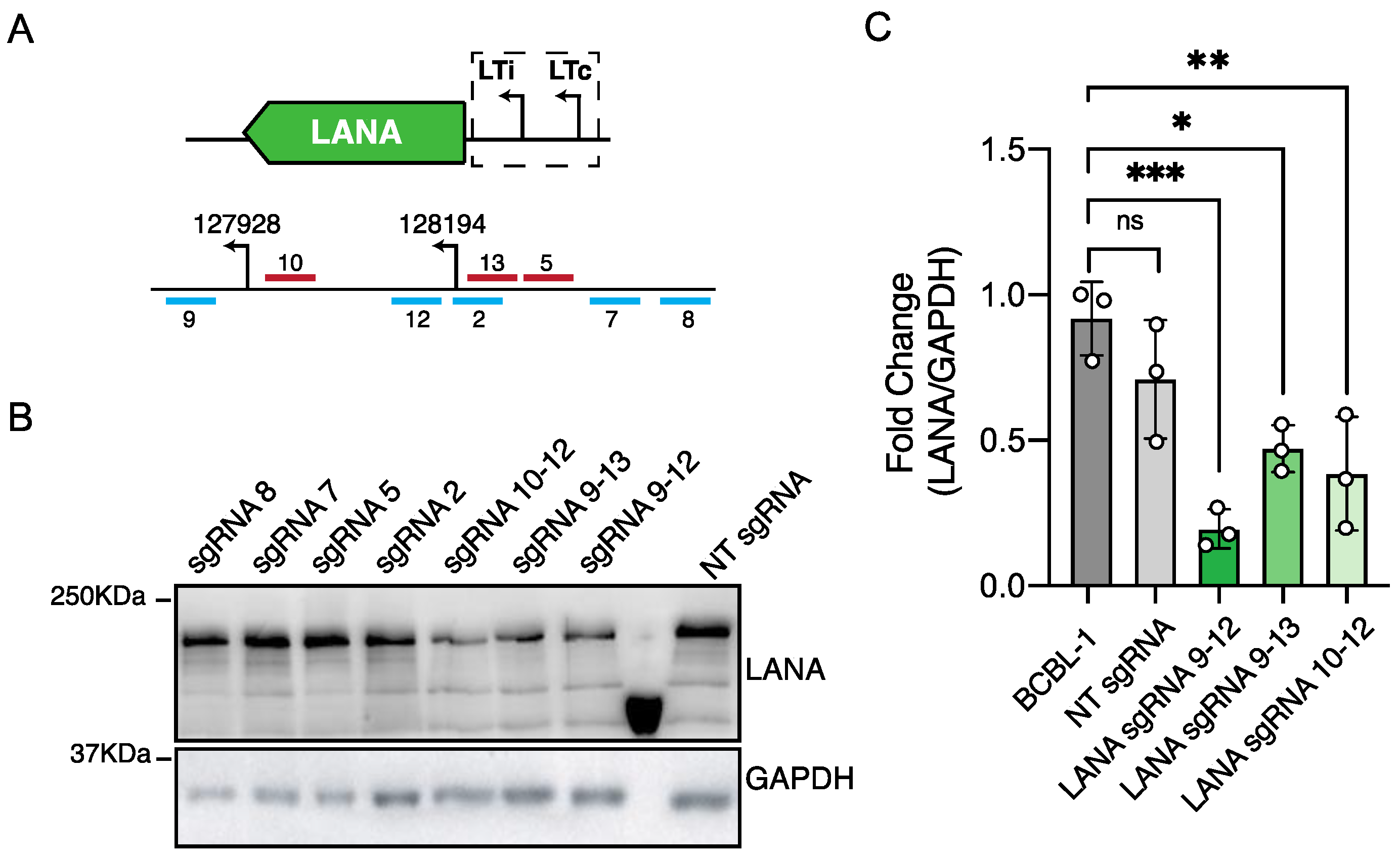
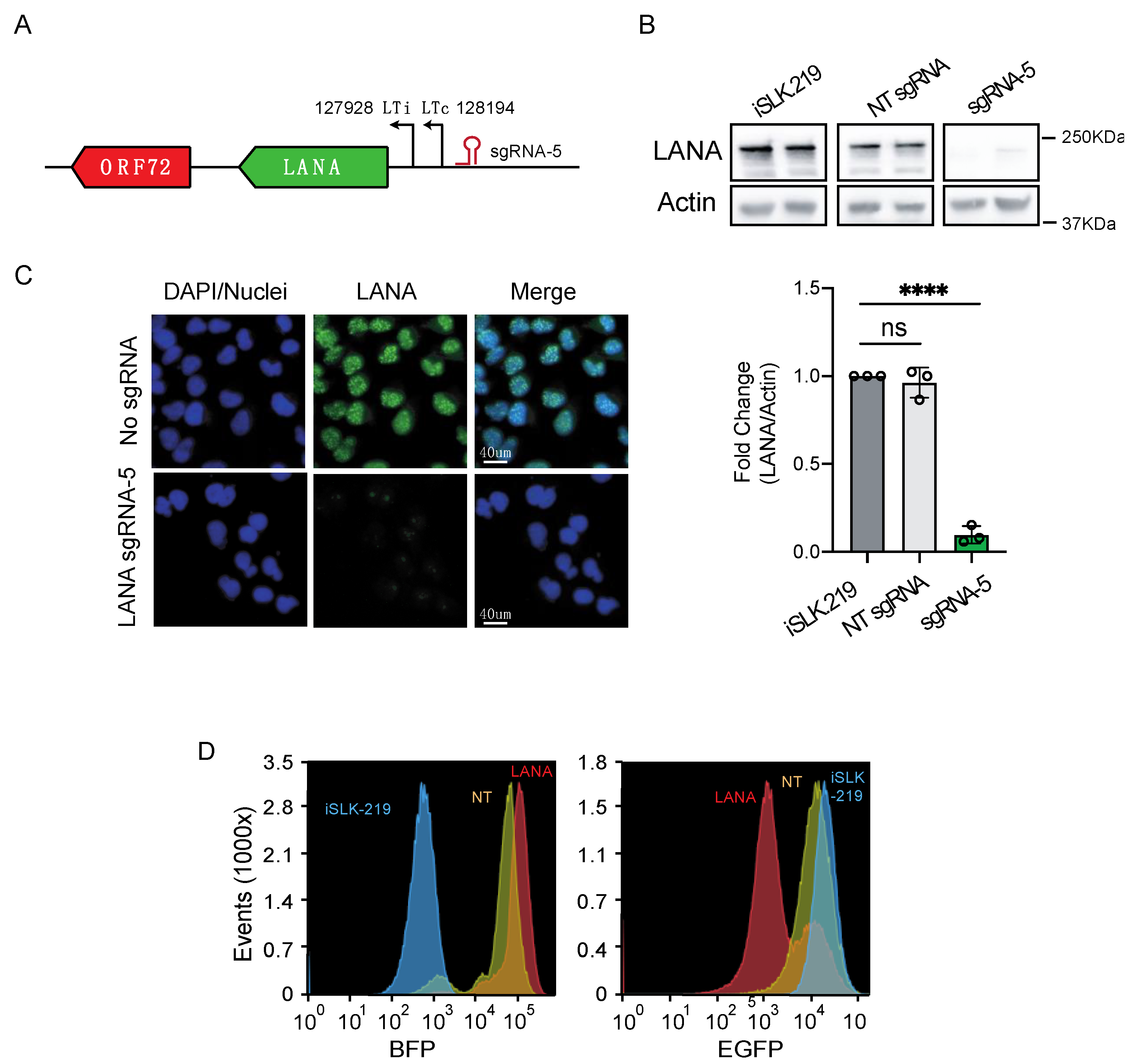
3.1. CRISPRi Represses Viral Genes in KSHV Infected Cells
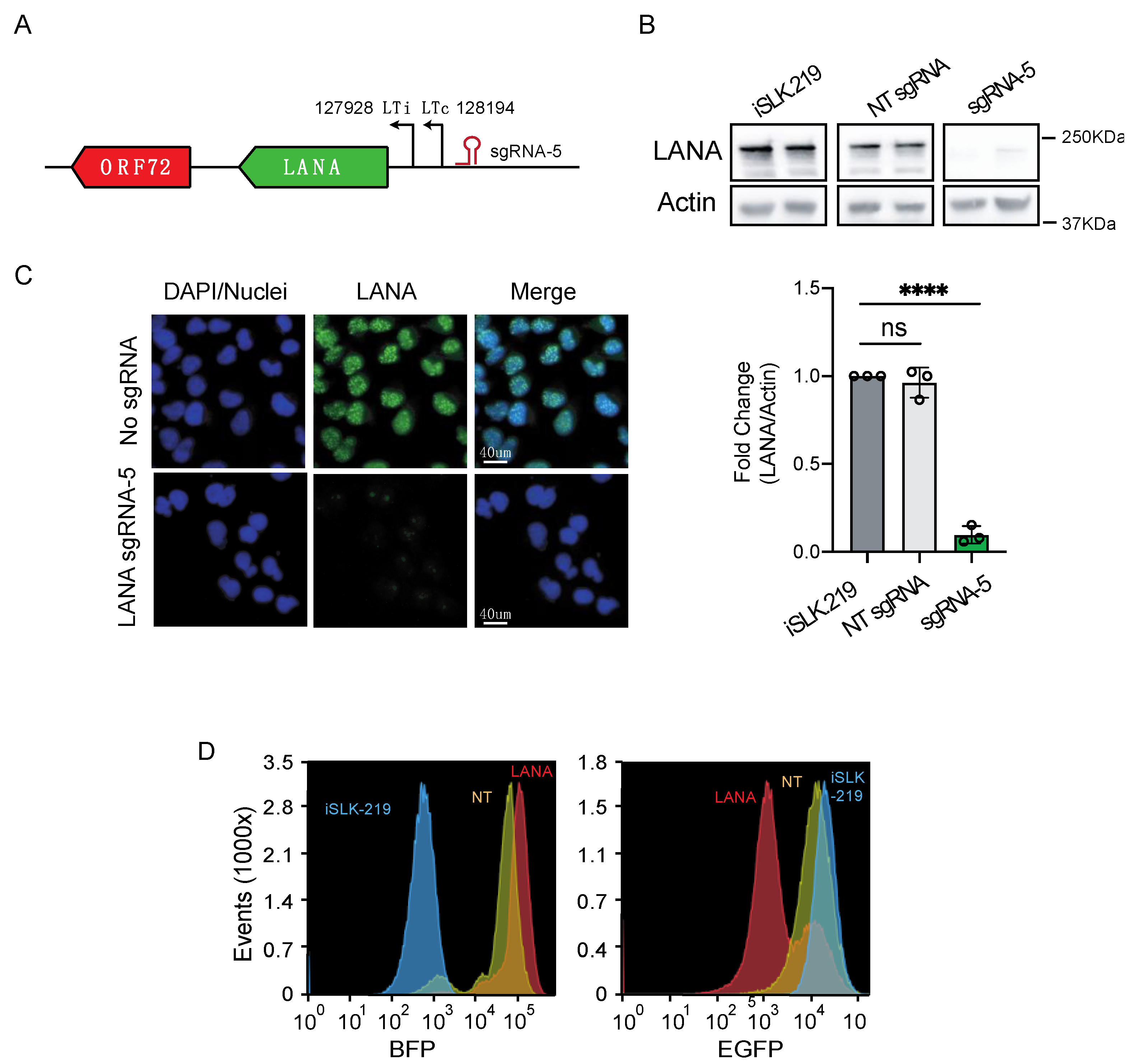
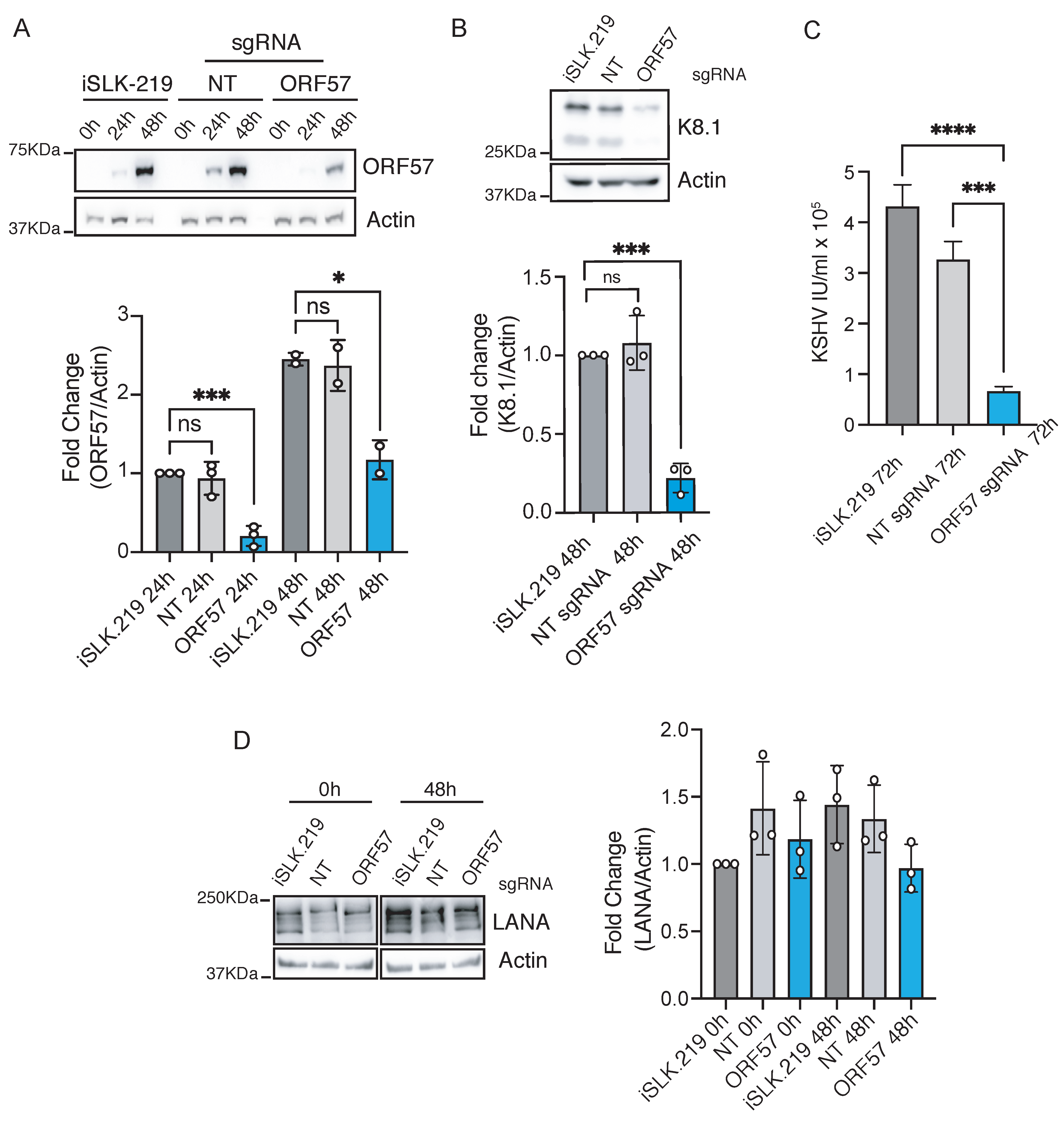
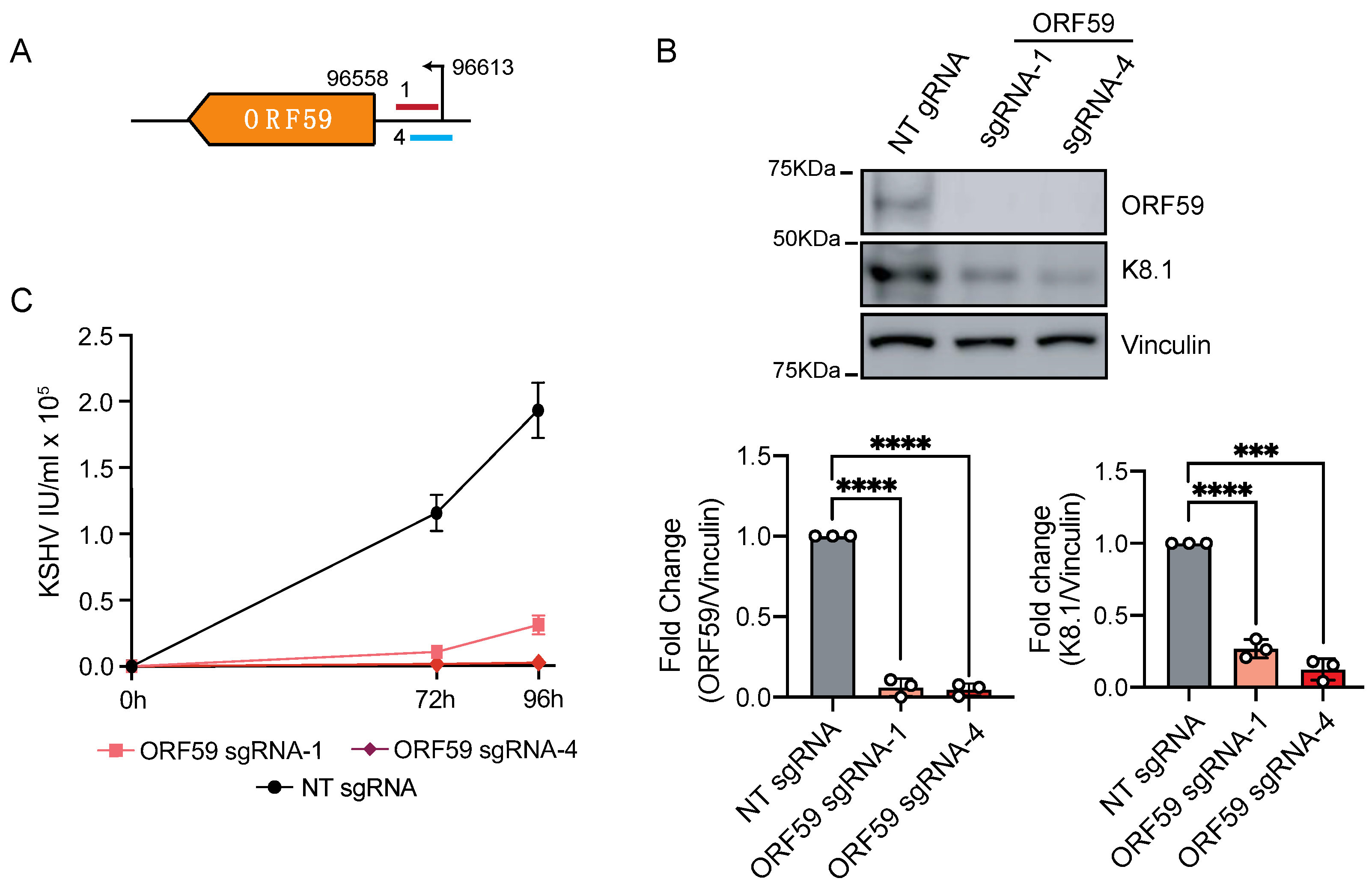
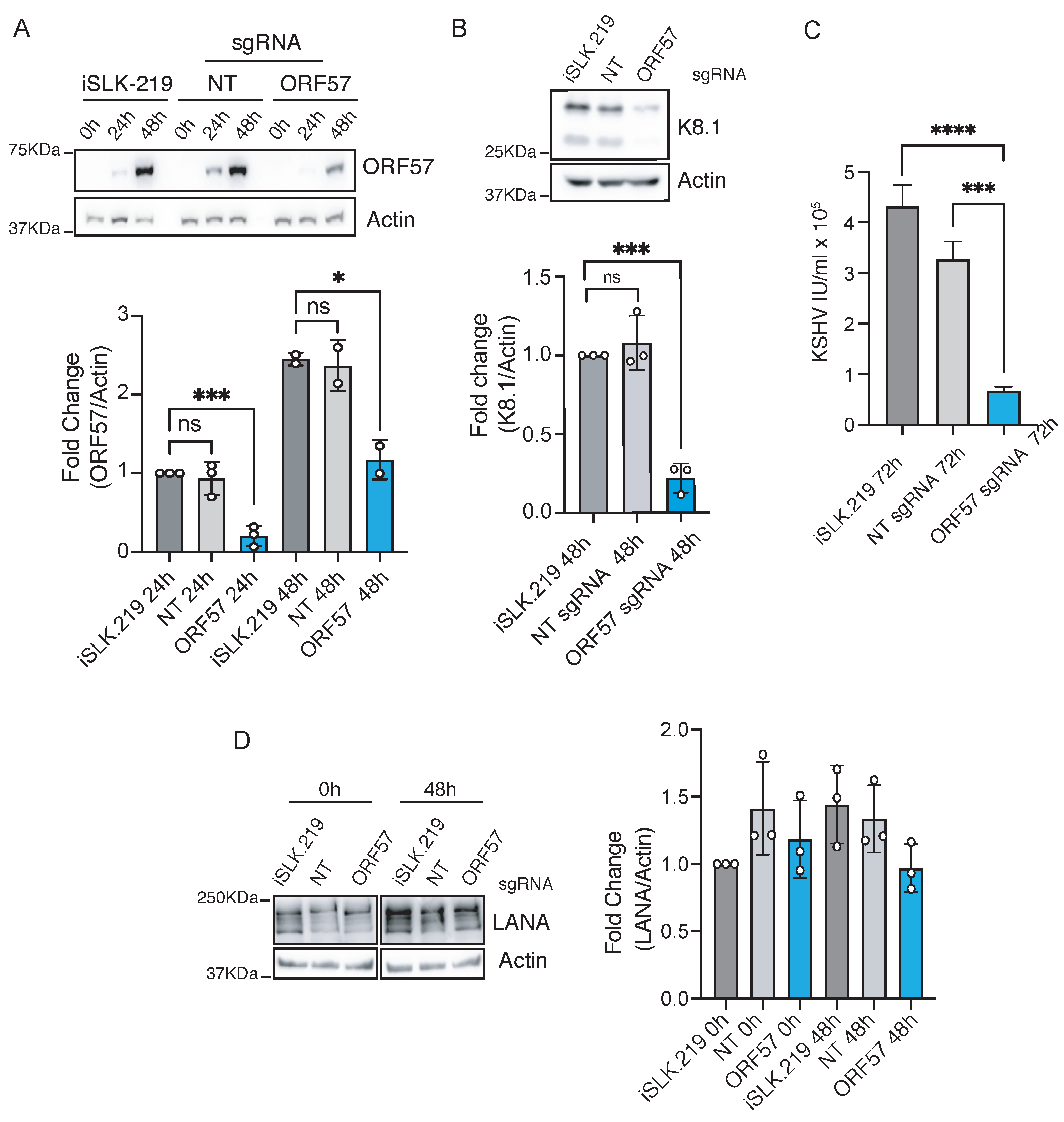
3.2. CRISPRi Represses Latent and Lytic KSHV Genes and Curtails Infectivity
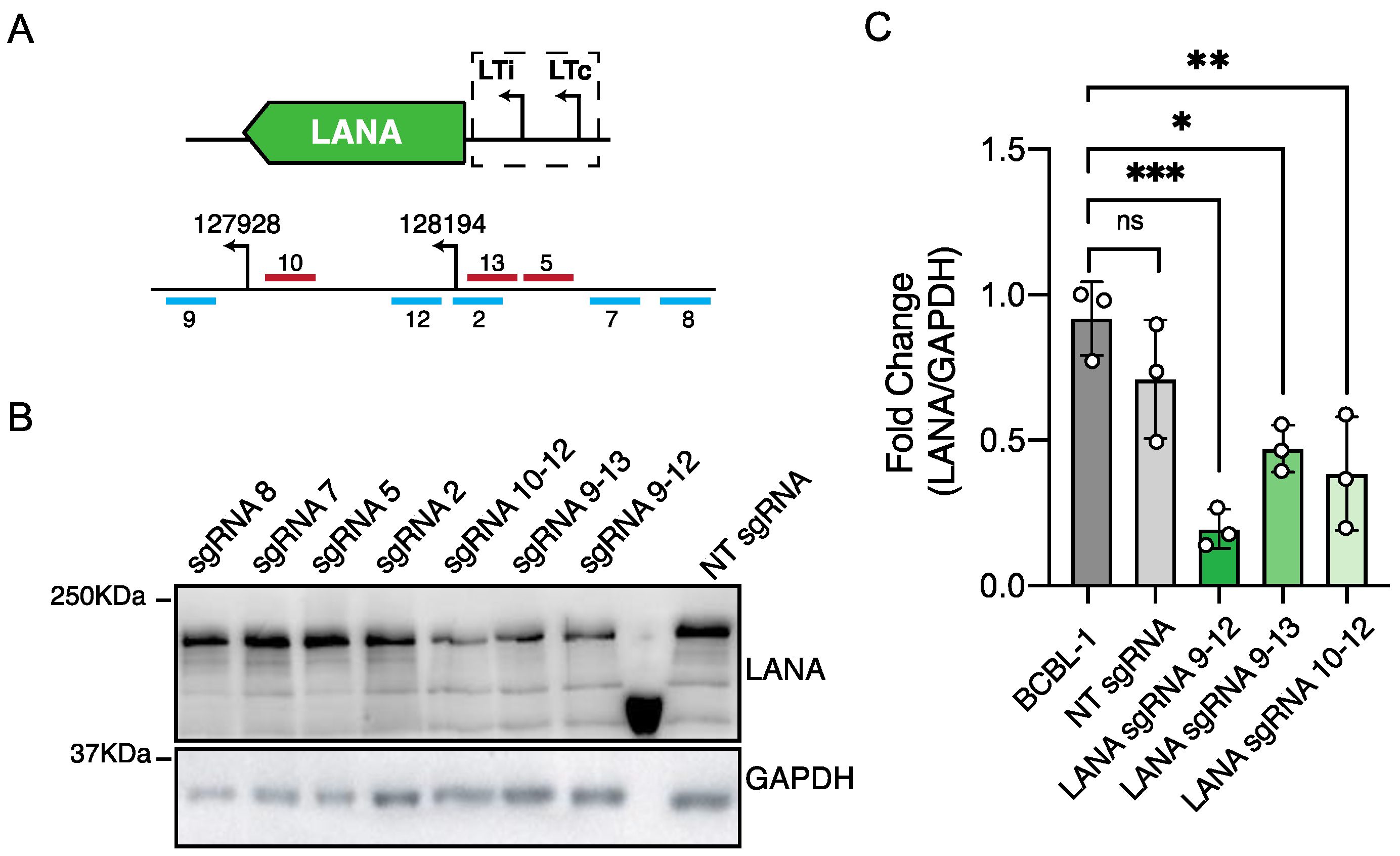
3.3. CRISPRi Represses Viral Genes in PEL-Derived Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dissinger, N.J.; Damania, B. Recent Advances in Understanding Kaposi’s Sarcoma-associated Herpesvirus. F1000Research 2016, 5, 740. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Globocan 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012; World Health Organisation (WHO): Geneva, Switzerland, 2015. [Google Scholar]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s Sarcoma and Its Associated Herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [Green Version]
- What Are the Key Statistics about Kaposi Sarcoma? American Cancer Society: Burlington, VT, USA, 2016.
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A Comprehensive Annotation of the Kaposi’s Sarcoma-Associated Herpesvirus Genome Using Next-Generation Sequencing Reveals Novel Genomic and Functional Features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhao, Y.; Karijolich, J. The Landscape of Transcription Initiation Across Latent and Lytic KSHV Genomes. PLoS Pathog. 2019, 15, e1007852. [Google Scholar] [CrossRef]
- Broussard, G.; Damania, B. KSHV: Immune Modulation and Immunotherapy. Front. Immunol. 2020, 10, 3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallin, L.E.; Goldschmidt-Clermont, P.; Mesri, E.A. Molecular and Cellular Mechanisms of KSHV Oncogenesis of Kaposi’s Sarcoma Associated with HIV/AIDS. PLoS Pathog. 2014, 10, e1004154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, B. Early Events in Kaposi’s Sarcoma-Associated Herpesvirus Infection of Target Cells. J. Virol. 2009, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, C.; Walsh, D.; Harbell, J.; Wilson, A.C.; Mohr, I. Activation of Host Translational Control Pathways by a Viral Developmental Switch. PLoS Pathog. 2009, 5, e1000334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacVeigh-Fierro, D.; Rodriguez, W.; Miles, J.; Muller, M. Stealing the Show: KSHV Hijacks Host RNA Regulatory Pathways to Promote Infection. Viruses 2020, 12, 1024. [Google Scholar] [CrossRef]
- Ohsaki, E.; Ueda, K. Interplay Between KSHV and the Host DNA Damage Response. Front. Cell. Infect. Microbiol. 2020, 10, 764. [Google Scholar] [CrossRef]
- Kumar, B.; Chandran, B. KSHV Entry and Trafficking in Target Cells–Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses 2016, 8, 305. [Google Scholar] [CrossRef]
- Zhou, F.C.; Zhang, Y.J.; Deng, J.H.; Wang, X.P.; Pan, H.Y.; Hettler, E.; Gao, S.J. Efficient Infection by a Recombinant Kaposi’s Sarcoma-Associated Herpesvirus Cloned in a Bacterial Artificial Chromosome: Application for Genetic Analysis. J. Virol. 2002, 76, 6185–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brulois, K.F.; Chang, H.; Lee, A.S.Y.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.-R.; Myoung, J.; Jung, J.U.; et al. Construction and Manipulation of a New Kaposi’s Sarcoma-Associated Herpesvirus Bacterial Artificial Chromosome Clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Qin, Z.; Riker, A.I.; Xi, Y. CRISPR/Cas9 Ablating Viral microRNA Promotes Lytic Reactivation of Kaposi’s Sarcoma-associated Herpesvirus. Biochem. Biophys. Res. Commun. 2020, 533, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Gabaev, I.; Williamson, J.C.; Crozier, T.W.; Schulz, T.F.; Lehner, P.J. Quantitative Proteomics Analysis of Lytic KSHV Infection in Human Endothelial Cells Reveals Targets of Viral Immune Modulation. Cell Rep. 2020, 33, 108249. [Google Scholar] [CrossRef] [PubMed]
- Tso, F.Y.; West, J.T.; Wood, C. Reduction of Kaposi’s Sarcoma-Associated Herpesvirus Latency Using CRISPR-Cas9 To Edit the Latency-Associated Nuclear Antigen Gene. J. Virol. 2019, 93, e02183-18. [Google Scholar] [CrossRef] [Green Version]
- Holmes, D.L.; Vogt, D.T.; Lagunoff, M. A CRISPR-Cas9 Screen Identifies Mitochondrial Translation as an Essential Process in Latent KSHV Infection of Human Endothelial Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28384–28392. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.G.; Nguyen, T.H.; Roberts, L.; Fischer, L.T.; Glickman, K.; Golas, G.; Papp, B.; Toth, Z. Epigenetic Factor siRNA Screen during Primary KSHV Infection Identifies Novel Host Restriction Factors for the Lytic Cycle of KSHV. PLoS Pathog. 2020, 16, e1008268. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.M.; Chau, B.N.; Cleary, M.A.; Lim, L.; Linsley, P.S. Widespread siRNA “Off-target” Transcript Silencing Mediated by Seed Region Sequence Complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.L.; Linsley, P.S. Recognizing and Avoiding siRNA Off-target Effects for Target Identification and Therapeutic Application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression Profiling Reveals Off-target Gene Regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- BeltCappellino, A.; Majerciak, V.; Lobanov, A.; Lack, J.; Cam, M.; Zheng, Z.-M. CRISPR/Cas9-Mediated Knockout and in situ Inversion of the ORF57 Gene from All Copies of the Kaposi’s Sarcoma-Associated Her-pesvirus Genome in BCBL-1 Cells. J. Virol. 2019, 93, e00628-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV Genome Replication and Maintenance. Front. Microbiol. 2016, 7, 54. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, R. KRAB-containing Zinc-finger Repressor Proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.D.; Suresh, S.; Schlecht, U.; Wu, M.; Wagih, O.; Peltz, G.; Davis, R.W.; Steinmetz, L.M.; Parts, L.; Onge, R.P. Quantitative CRISPR Interference Screens in Yeast Identify Chemical-genetic Interactions and New Rules for Guide RNA Design. Genome Biol. 2016, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Todor, H.; Silvis, M.R.; Osadnik, H.; Gross, C.A. Bacterial CRISPR Screens for Gene Function. Curr. Opin. Microbiol. 2021, 59, 102–109. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Wong, S.; Peters, J.M.; Almeida, R.; Qi, L.S. Targeted Transcriptional Repression in Bacteria Using CRISPR Interference (CRISPRi). Methods Mol. Biol. 2015, 1311, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Kampmann, M.; et al. Paradoxical Resistance of Multiple Myeloma to Proteasome Inhibitors by Decreased Levels of 19S Pro-teasomal Subunits. Elife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Fields, A.P.; Park, C.Y.; Corn, J.E.; Weissman, J.S.; et al. Compact and Highly Active Next-generation Libraries for CRISPR-mediated Gene Repression and Activation. Elife 2016, 5, e19760. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Renne, R.; Ganem, D.; Forghani, B. Human Herpesvirus 8 Glycoprotein K8.1: Expression, Post-translational Modification and Localization Analyzed by Monoclonal Antibody. J. Clin. Virol. 2000, 17, 127–136. [Google Scholar] [CrossRef]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic Growth of Kaposi’s Sarcoma–associated Herpesvirus (Human Herpesvirus 8) in Culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef]
- Myoung, J.; Ganem, D. Generation of a Doxycycline-inducible KSHV Producer Cell Line of Endothelial Origin: Maintenance of Tight Latency with Efficient Reactivation upon Induction. J. Virol. Methods 2011, 174, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Vieira, J.; O’Hearn, P.M. Use of the Red Fluorescent Protein as a Marker of Kaposi’s Sarcoma-associated Herpesvirus Lytic Gene Expression. Virology 2004, 325, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular Biology of KSHV Lytic Reactivation. Viruses 2015, 7, 116–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Ballestas, M.E.; Kaye, K.M. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 Mediates Episome Persistence through cis Acting Terminal Repeat (TR) Sequence and Specifically Binds TR DNA. J. Virol. 2001, 75, 3250–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient Persistence of Extrachromosomal KSHV DNA Mediated by Laten-cy-Associated Nuclear Antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Garber, A.C.; Renne, R. The Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpes-virus Supports Latent DNA Replication in Dividing Cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, T.F.; Chang, Y. KSHV Gene Expression and Regulation. In Human Herpesviruses; Cambridge University Press (CUP): Cambridge, UK, 2010; pp. 490–513. [Google Scholar]
- Majerciak, V.; Zheng, Z.-M. KSHV ORF57, a Protein of Many Faces. Viruses 2015, 7, 604–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerciak, V.; Pripuzova, N.; McCoy, J.P.; Gao, S.-J.; Zheng, Z.-M. Targeted Disruption of Kaposi’s Sar-coma-Associated Herpesvirus ORF57 in the Viral Genome Is Detrimental for the Expression of ORF59, K8α, and K8.1 and the Production of Infectious Virus. J. Virol. 2007, 81, 1062–1071. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.R.; Chandran, B. Characterization of Human Herpesvirus 8 ORF59 Protein (PF-8) and Mapping of the Proces-sivity and Viral DNA Polymerase-Interacting Domains. J. Virol. 2000, 74, 10920–10929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papousková, I.; Tarrant, M.; Pari, G.S. Amplification of the Kaposi’s Sarcoma-associated Herpesvirus/Human Herpesvirus 8 Lytic Origin of DNA Replication is Dependent upon a Cis-acting AT-rich Region and an ORF50 Response Element and the Trans-acting Factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xulin, C.; Ricciardi, R.P. Human Kaposi’s Sarcoma Herpesvirus Processivity Factor-8 Functions as a Dimer in DNA Synthesis. J. Biol. Chem. 2004, 279, 28375–28386. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.; Matsumura, S.; Wilson, A.C. Transcripts Encoding K12, v-FLIP, v-Cyclin, and the MicroRNA Cluster of Kaposi’s Sarcoma-Associated Herpesvirus Originate from a Common Promoter. J. Virol. 2005, 79, 14457–14464. [Google Scholar] [CrossRef] [Green Version]
- Hein, M.Y.; Weissman, J.S. Functional Single-cell Genomics of Human Cytomegalovirus Infection. bioRxiv 2019, 775080. [Google Scholar]
- Radzisheuskaya, A.; Shlyueva, D.; Müller, I.; Helin, K. Optimizing sgRNA Position Markedly Improves the Efficiency of CRISPR/dCas9-mediated Transcriptional Repression. Nucleic Acids Res. 2016, 44, e141. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, S.; Fujita, Y.; Gomez, E.; Tanese, N.; Wilson, A.C. Activation of the Kaposi’s Sarcoma-Associated Herpesvirus Major Latency Locus by the Lytic Switch Protein RTA (ORF50). J. Virol. 2005, 79, 8493–8505. [Google Scholar] [CrossRef] [Green Version]
- Elbasani, E.; Falasco, F.; Gramolelli, S.; Nurminen, V.; Günther, T.; Weltner, J.; Balboa, D.; Grundhoff, A.; Otonkoski, T.; Ojala, P.M. Kaposi’s Sarcoma-Associated Herpesvirus Reactivation by Targeting of a dCas9-Based Transcription Ac-tivator to the ORF50 Promoter. Viruses 2020, 12, 952. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brackett, K.; Mungale, A.; Lopez-Isidro, M.; Proctor, D.A.; Najarro, G.; Arias, C. CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells. Viruses 2021, 13, 783. https://doi.org/10.3390/v13050783
Brackett K, Mungale A, Lopez-Isidro M, Proctor DA, Najarro G, Arias C. CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells. Viruses. 2021; 13(5):783. https://doi.org/10.3390/v13050783
Chicago/Turabian StyleBrackett, Kevin, Ameera Mungale, Mary Lopez-Isidro, Duncan A. Proctor, Guillermo Najarro, and Carolina Arias. 2021. "CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells" Viruses 13, no. 5: 783. https://doi.org/10.3390/v13050783
APA StyleBrackett, K., Mungale, A., Lopez-Isidro, M., Proctor, D. A., Najarro, G., & Arias, C. (2021). CRISPR Interference Efficiently Silences Latent and Lytic Viral Genes in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Cells. Viruses, 13(5), 783. https://doi.org/10.3390/v13050783