
The Translated Amino Acid Sequence of an Insertion in the Hepatitis E Virus Strain 47832c Genome, But Not the RNA Sequence, Is Essential for Efficient Cell Culture Replication


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Generation of Plasmids Carrying Deletions and Sequence Substitutions in the 47832c Genome
2.3. Generation of Infectious Viruses from Plasmids
2.4. Immunofluorescence Assay
2.5. Reverse Transcription—Quantitative PCR (RT-qPCR)
2.6. Generation of Persistently Infected Cell Lines
2.7. Viral Growth Kinetics
2.8. Statististical Analyses
3. Results
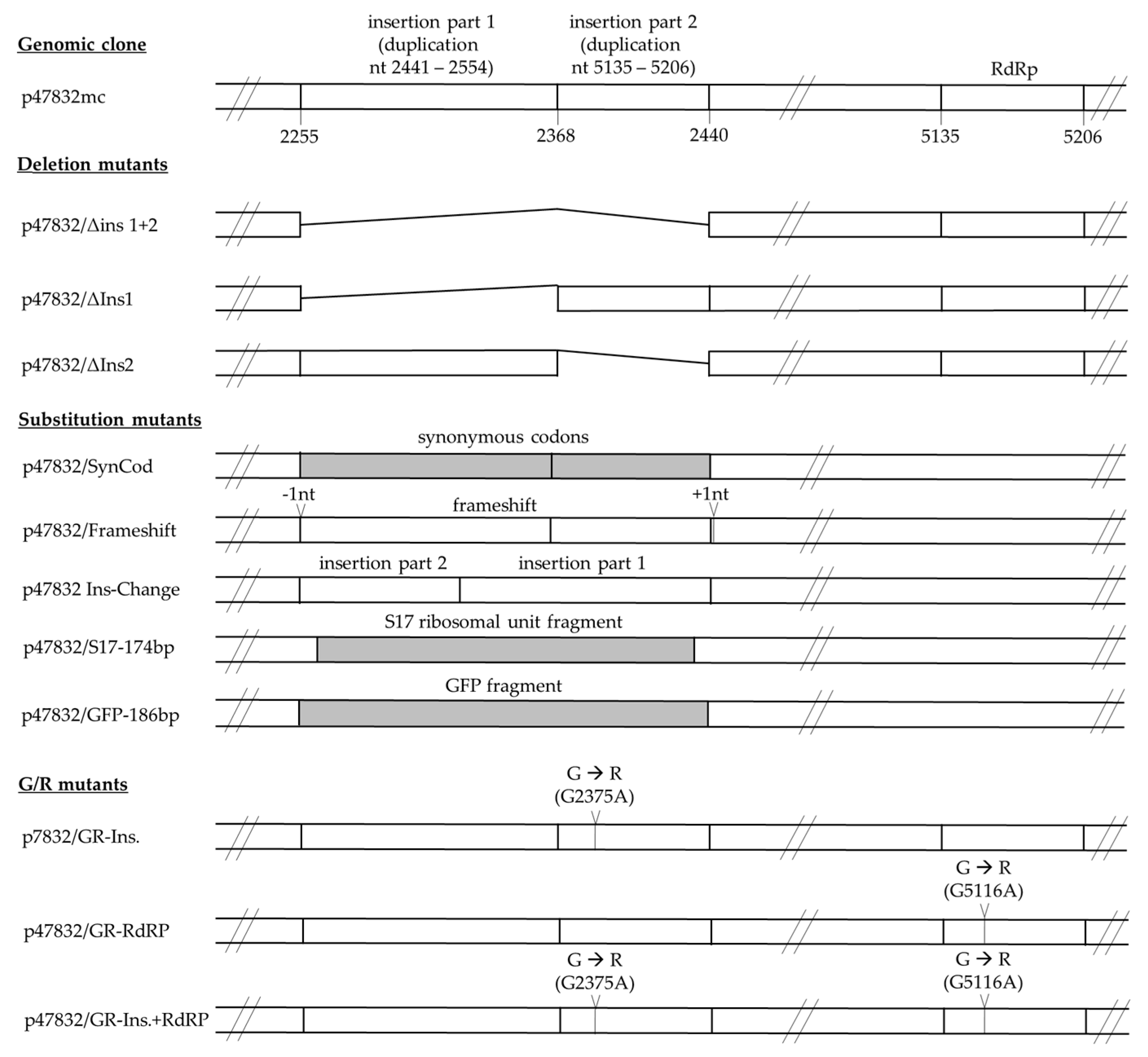
3.1. Generation of Constructs Containing Deletions or Substitutions in the Insertion of p47832mc
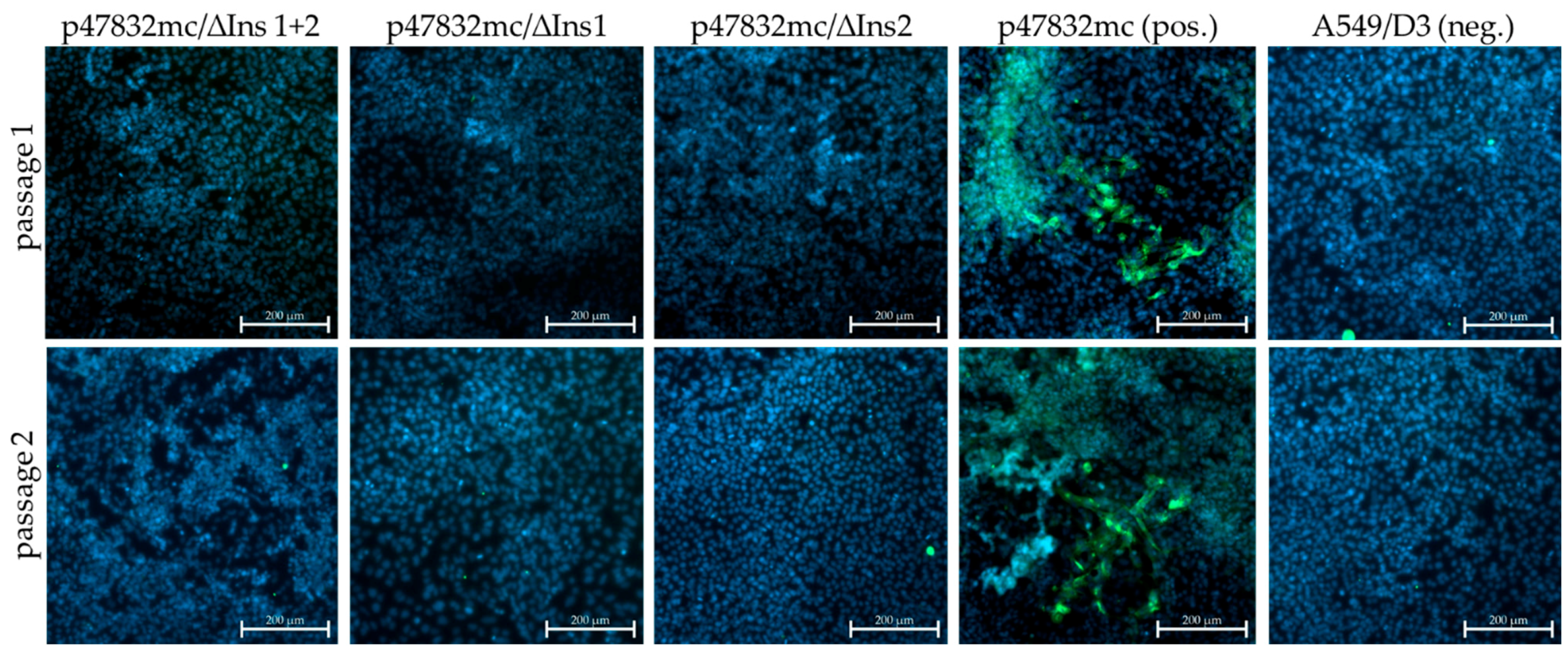
3.2. Effect of Deletions within the Insertion Region of the HVR
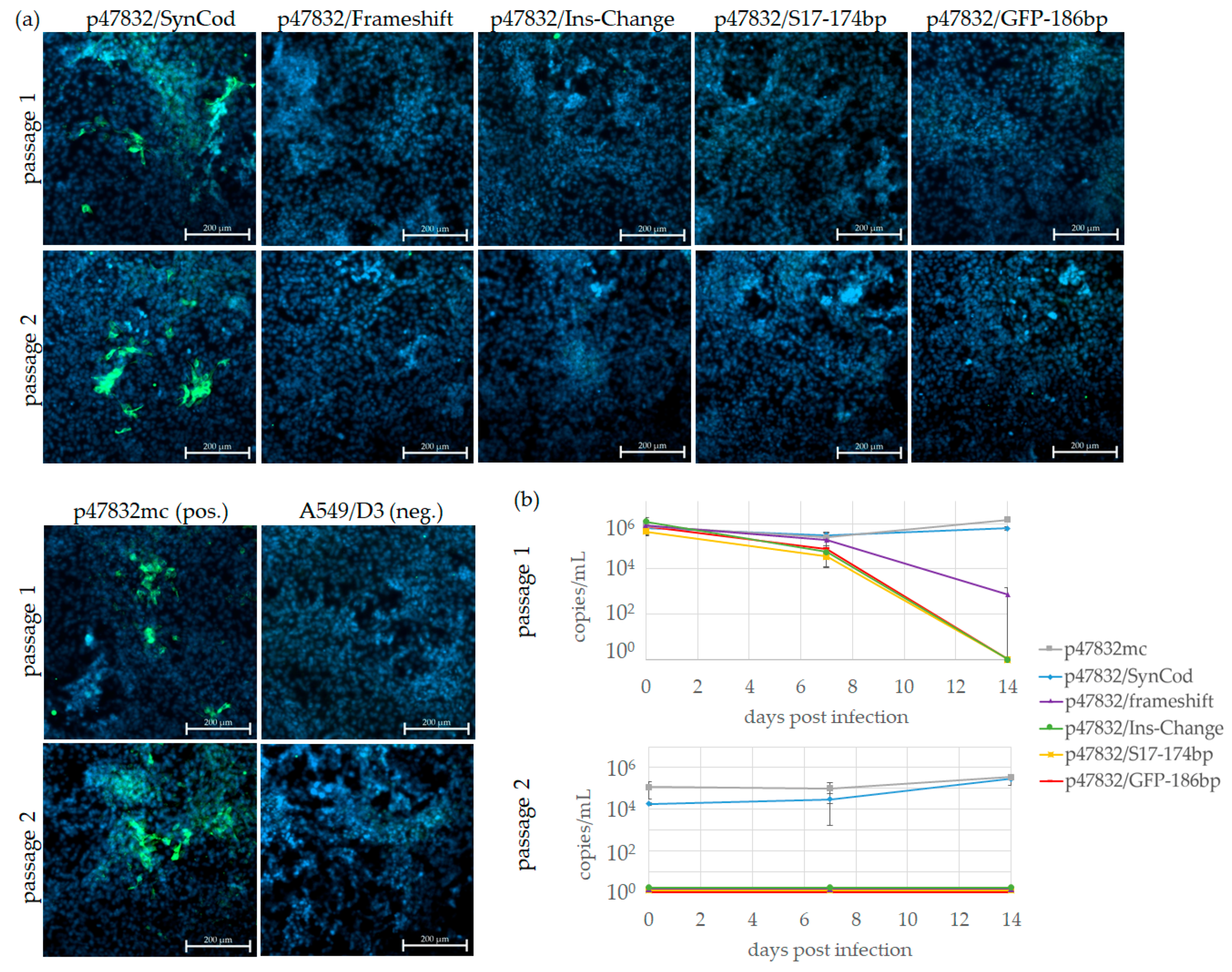
3.3. Effect of Substitutions of the Insertion Region of the HVR
3.4. Influence of Specific Glycine-to-Arginine Mutations on Virus Replication
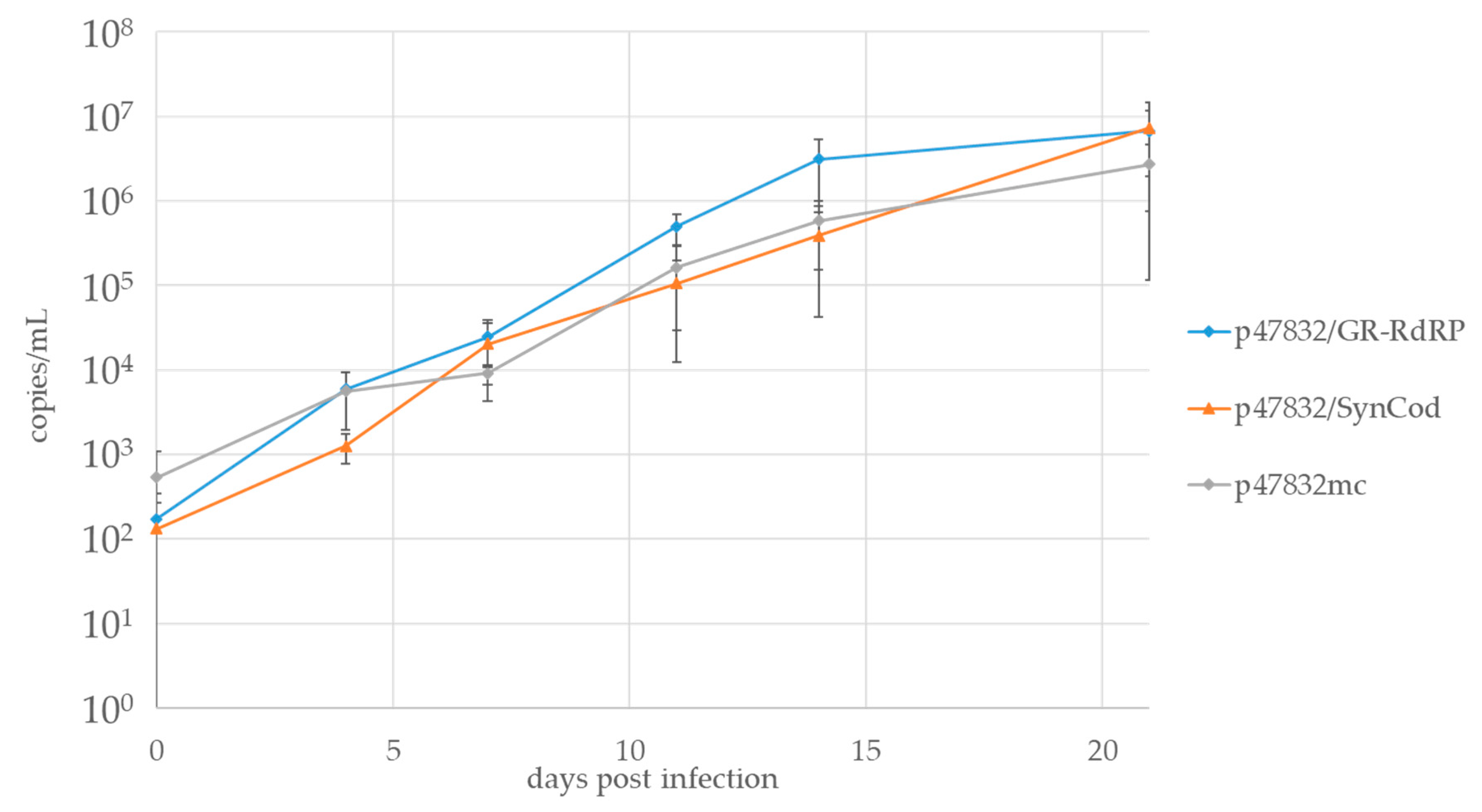
3.5. Comparison of Growth Kinetics of p47832/GR-RdRp, p47832/SynCod and p47832mc
3.6. Testing the Capability of the Constructs to Generate Persistently Virus-Infected Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamar, N.; Bendall, R.; Legrand-Abravanel, F.; Xia, N.S.; Ijaz, S.; Izopet, J.; Dalton, H.R. Hepatitis E. Lancet 2012, 379, 2477–2488. [Google Scholar] [CrossRef]
- Sayed, I.M.; El-Mokhtar, M.A.; Mahmoud, M.A.R.; Elkhawaga, A.A.; Gaber, S.; Seddek, N.H.; Abdel-Wahid, L.; Ashmawy, A.M.; Alkareemy, E.A.R. Clinical Outcomes and Prevalence of Hepatitis E Virus (HEV) Among Non-A-C Hepatitis Patients in Egypt. Infect. Drug Resist. 2021, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Khuroo, M.S.; Khuroo, N.S. Transmission of Hepatitis E Virus in Developing Countries. Viruses 2016, 8, 253. [Google Scholar] [CrossRef]
- Lu, L.; Li, C.; Hagedorn, C.H. Phylogenetic analysis of global hepatitis E virus sequences: Genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 2006, 16, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.T.; Balaban, H.Y. Hepatitis E virus: Epidemiology, diagnosis, clinical manifestations, and treatment. World J. Gastroenterol. 2020, 26, 5543–5560. [Google Scholar] [CrossRef]
- Adlhoch, C.; Avellon, A.; Baylis, S.A.; Ciccaglione, A.R.; Couturier, E.; de Sousa, R.; Epstein, J.; Ethelberg, S.; Faber, M.; Feher, A.; et al. Hepatitis E virus: Assessment of the epidemiological situation in humans in Europe, 2014/15. J. Clin. Virol. 2016, 82, 9–16. [Google Scholar] [CrossRef]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef]
- Pallerla, S.R.; Harms, D.; Johne, R.; Todt, D.; Steinmann, E.; Schemmerer, M.; Wenzel, J.J.; Hofmann, J.; Shih, J.W.K.; Wedemeyer, H.; et al. Hepatitis E Virus Infection: Circulation, Molecular Epidemiology, and Impact on Global Health. Pathogens 2020, 9, 856. [Google Scholar] [CrossRef]
- Oechslin, N.; Moradpour, D.; Gouttenoire, J. On the Host Side of the Hepatitis E Virus Life Cycle. Cells 2020, 9, 1294. [Google Scholar] [CrossRef]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Dao Thi, V.L.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 2018, 14, e1007471. [Google Scholar] [CrossRef] [PubMed]
- Jameel, S.; Zafrullah, M.; Ozdener, M.H.; Panda, S.K. Expression in animal cells and characterization of the hepatitis E virus structural proteins. J. Virol. 1996, 70, 207–216. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A. Evolution of the hepatitis E virus polyproline region: Order from disorder. J. Virol. 2012, 86, 10186–10193. [Google Scholar] [CrossRef]
- Purdy, M.A.; Lara, J.; Khudyakov, Y.E. The hepatitis E virus polyproline region is involved in viral adaptation. PLoS ONE 2012, 7, e35974. [Google Scholar] [CrossRef]
- Pudupakam, R.S.; Kenney, S.P.; Cordoba, L.; Huang, Y.W.; Dryman, B.A.; Leroith, T.; Pierson, F.W.; Meng, X.J. Mutational analysis of the hypervariable region of hepatitis e virus reveals its involvement in the efficiency of viral RNA replication. J. Virol. 2011, 85, 10031–10040. [Google Scholar] [CrossRef]
- Munoz-Chimeno, M.; Cenalmor, A.; Garcia-Lugo, M.A.; Hernandez, M.; Rodriguez-Lazaro, D.; Avellon, A. Proline-Rich Hypervariable Region of Hepatitis E Virus: Arranging the Disorder. Microorganisms 2020, 8, 1417. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Nicot, F.; Jeanne, N.; Dimeglio, C.; Roulet, A.; Lefebvre, C.; Carcenac, R.; Manno, M.; Dubois, M.; Peron, J.M.; et al. Insertions and Duplications in the Polyproline Region of the Hepatitis E Virus. Front. Microbiol. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef]
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J. Virol. 2012, 86, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Trojnar, E.; Filter, M.; Hofmann, J. Thermal Stability of Hepatitis E Virus as Estimated by a Cell Culture Method. Appl. Environ. Microbiol. 2016, 82, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Wolff, A.; Gadicherla, A.K.; Filter, M.; Schluter, O. Stability of hepatitis E virus at high hydrostatic pressure processing. Int. J. Food Microbiol. 2021, 339, 109013. [Google Scholar] [CrossRef]
- Wolff, A.; Gunther, T.; Albert, T.; Johne, R. Effect of Sodium Chloride, Sodium Nitrite and Sodium Nitrate on the Infectivity of Hepatitis E Virus. Food Environ. Virol. 2020, 12, 350–354. [Google Scholar] [CrossRef]
- Wolff, A.; Gunther, T.; Albert, T.; Schilling-Loeffler, K.; Gadicherla, A.K.; Johne, R. Stability of hepatitis E virus at different pH values. Int. J. Food Microbiol. 2020, 325, 108625. [Google Scholar] [CrossRef] [PubMed]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Johne, R.; Reuter, A.; Spiric, J.; Schwaben, L.; Grunweller, A.; Hildt, E. Inhibition of Hepatitis E Virus Spread by the Natural Compound Silvestrol. Viruses 2018, 10, 301. [Google Scholar] [CrossRef]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Schollmeier, A.; Johne, R.; Praefcke, G.J.K.; Hildt, E. Identification of the interferon-inducible GTPase GBP1 as major restriction factor for the Hepatitis E virus. J. Virol. 2021. [Google Scholar] [CrossRef]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, e15–e16. [Google Scholar] [CrossRef]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Bachlein, C.; Gadicherla, A.K.; Falkenhagen, A.; Tausch, S.H.; Johne, R. Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c. Pathogens 2020, 9, 157. [Google Scholar] [CrossRef]
- Schemmerer, M.; Apelt, S.; Trojnar, E.; Ulrich, R.G.; Wenzel, J.J.; Johne, R. Enhanced Replication of Hepatitis E Virus Strain 47832c in an A549-Derived Subclonal Cell Line. Viruses 2016, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [CrossRef]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef]
- Schielke, A.; Filter, M.; Appel, B.; Johne, R. Thermal stability of hepatitis E virus assessed by a molecular biological approach. Virol. J. 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Bruggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct * | 1st Passage (A549/D3 Cells) | 2nd Passage (A549/D3 Cells) | Persistently Infected A549/D3 Cell Line |
|---|---|---|---|
| p47832mc | + | + | + |
| p47832/Δins 1+2 | - | - | - |
| p47832/ΔIns1 | - | - | - |
| p47832/ΔIns2 | - | - | - |
| p47832/SynCod | + | + | + |
| p47832/Frameshift | - | - | - |
| p47832/Ins-Change | - | - | - |
| p47832/S17-174bp | - | - | - |
| p47832/GFP-186bp | - | - | - |
| p47832/GR-Ins | + | - | + |
| p47832/GR-RdRp | + | + | + |
| p47832/GR-Ins+RdRp | + | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholz, J.; Falkenhagen, A.; Johne, R. The Translated Amino Acid Sequence of an Insertion in the Hepatitis E Virus Strain 47832c Genome, But Not the RNA Sequence, Is Essential for Efficient Cell Culture Replication. Viruses 2021, 13, 762. https://doi.org/10.3390/v13050762
Scholz J, Falkenhagen A, Johne R. The Translated Amino Acid Sequence of an Insertion in the Hepatitis E Virus Strain 47832c Genome, But Not the RNA Sequence, Is Essential for Efficient Cell Culture Replication. Viruses. 2021; 13(5):762. https://doi.org/10.3390/v13050762
Chicago/Turabian StyleScholz, Johannes, Alexander Falkenhagen, and Reimar Johne. 2021. "The Translated Amino Acid Sequence of an Insertion in the Hepatitis E Virus Strain 47832c Genome, But Not the RNA Sequence, Is Essential for Efficient Cell Culture Replication" Viruses 13, no. 5: 762. https://doi.org/10.3390/v13050762
APA StyleScholz, J., Falkenhagen, A., & Johne, R. (2021). The Translated Amino Acid Sequence of an Insertion in the Hepatitis E Virus Strain 47832c Genome, But Not the RNA Sequence, Is Essential for Efficient Cell Culture Replication. Viruses, 13(5), 762. https://doi.org/10.3390/v13050762