
Screening of Bacteriophage Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Bacteriophages and Culture Media
2.2. Phage Isolation and Purification
2.3. Electron Microscopy
2.4. Host Range Determination
2.5. Genome Sequencing and Analysis
2.6. Proteomics
2.7. DNA Methods
2.8. Bioinformatics
2.9. Protein Function and Sequence Analysis
2.10. Confirmation of Toxicity by Growth Curve Analysis
3. Results
3.1. fHy-Eco03 Host Range and Growth Characteristics
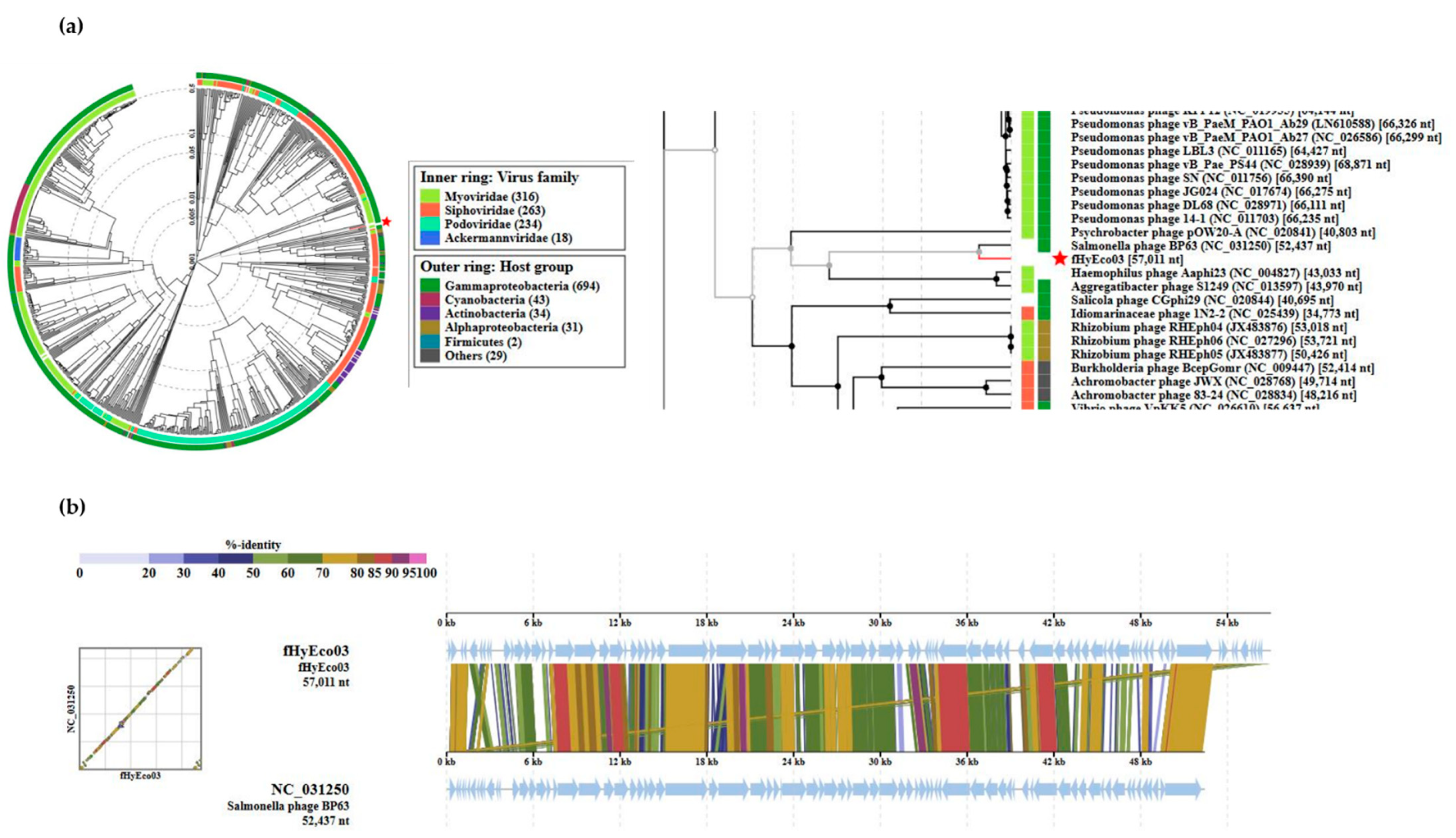
3.2. General Genomic Features of fHy-Eco03
3.3. Identification of toxHPUFs by Plating Efficiency and NGS-Based Screening Assays
3.4. Screening the fHy-Eco03-Encoded HPUF Genes
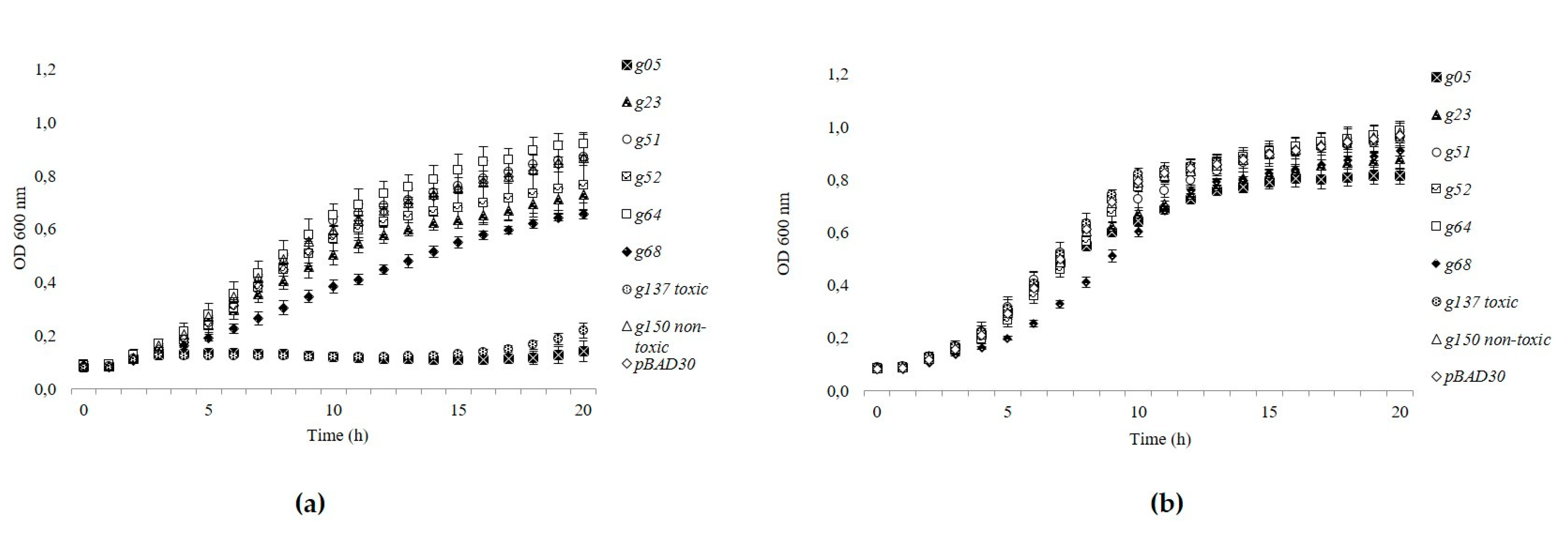
3.5. Gp05 Showed Toxicity among the Tested toxHPUF Candidates of fHy-Eco03
3.6. Functional and Structural Analysis of Gp05
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.B.; Brass, E.P.; Gilbert, D.N.; Bartlett, J.G.; Spellberg, B. Sustainable discovery and development of antibiotics—Is a nonprofit approach the future? N. Engl. J. Med. 2019, 381, 503–505. [Google Scholar] [CrossRef]
- Endersen, L.; O’Mahony, J.; Hill, C.; Ross, R.P.; McAuliffe, O.; Coffey, A. Phage therapy in the food industry. Annu. Rev. Food Sci. Technol. 2014, 5, 327–349. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, B.L. Holins in bacteria, eukaryotes, and archaea: Multifunctional xenologues with potential biotechnological and biomedical applications. J. Bacteriol. 2015, 197, 7–17. [Google Scholar] [CrossRef]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins: Applications for food safety. Curr. Opin. Biotechnol. 2016, 37, 76–87. [Google Scholar] [CrossRef]
- Liu, J.; Dehbi, M.; Moeck, G.; Arhin, F.; Bauda, P.; Bergeron, D.; Callejo, M.; Ferretti, V.; Ha, N.; Kwan, T.; et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004, 22, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, Y.; Dabbs, E.R. Phage as a source of antibacterial genes: Multiple inhibitory products encoded by Rhodococcus phage YF1. Bacteriophage 2011, 1, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, U.; Wan, X.; Spruit, C.M.; Skurnik, M.; Pajunen, M.I. A toxicity screening approach to identify bacteriophage-encoded anti-microbial proteins. Viruses 2019, 11, 1057. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Godavarthi, S.; Kumar, A.; Sen, R. A mycobacteriophage genomics approach to identify novel mycobacteriophage proteins with mycobactericidal properties. Microbiology 2019, 165, 722–736. [Google Scholar] [CrossRef]
- Spruit, C.M.; Wicklund, A.; Wan, X.; Skurnik, M.; Pajunen, M.I. Discovery of three toxic proteins of Klebsiella phage fHe-Kpn01. Viruses 2020, 12, 544. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, A.; Ceyssens, P.J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.S.; Cenens, W.; Aertsen, A.; et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef]
- Lamberg, A.; Nieminen, S.; Qiao, M.; Savilahti, H. Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl. Environ. Microb. 2002, 68, 705–712. [Google Scholar] [CrossRef]
- Leon-Velarde, C.G.; Happonen, L.; Pajunen, M.; Leskinen, K.; Kropinski, A.M.; Mattinen, L.; Rajtor, M.; Zur, J.; Smith, D.; Chen, S.; et al. Yersinia enterocolitica-specific infection by bacteriophages TG1 and φR1-RT is dependent on temperature-regulated expression of the phage host receptor OmpF. Appl. Environ. Microb. 2016, 82, 5340–5353. [Google Scholar] [CrossRef]
- Grant, S.G.; Jessee, J.; Bloom, F.R.; Hanahan, D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 1990, 87, 4645–4649. [Google Scholar] [CrossRef]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Sambrook, J.; Russell, D.W. Purification of bacteriophage lambda particles by centrifugation through a glycerol step gradient. CSH Protoc. 2006, 2006, pdb–prot3969. [Google Scholar] [PubMed]
- Ackermann, H.W. Basic phage electron microscopy. Methods Mol. Biol. 2009, 501, 113–126. [Google Scholar] [PubMed]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Goker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Gambino, M.; Norgaard Sorensen, A.; Ahern, S.; Smyrlis, G.; Gencay, Y.E.; Hendrix, H.; Neve, H.; Noben, J.P.; Lavigne, R.; Brondsted, L. Phage S144, a new polyvalent phage infecting Salmonella spp. and Cronobacter sakazakii. Int. J. Mol. Sci. 2020, 21, 5196. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Scaife, S.; Raven, N.D.; Wigley, D.B. Crystallization and preliminary X-ray analysis of RecG, a replication-fork reversal helicase from Thermotoga maritima complexed with a three-way DNA junction. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1695–1696. [Google Scholar] [CrossRef]
- Lloyd, R.G.; Rudolph, C.J. 25 years on and no end in sight: A perspective on the role of RecG protein. Curr. Genet. 2016, 62, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.J.; Mahdi, A.A.; Upton, A.L.; Lloyd, R.G. RecG protein and single-strand DNA exonucleases avoid cell lethality associated with PriA helicase activity in Escherichia coli. Genetics 2010, 186, 473–492. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Upton, A.L.; Briggs, G.S.; Lloyd, R.G. Is RecG a general guardian of the bacterial genome? DNA Repair 2010, 9, 210–223. [Google Scholar] [CrossRef]
- Ruckman, J.; Ringquist, S.; Brody, E.; Gold, L. The bacteriophage T4 regB ribonuclease. Stimulation of the purified enzyme by ribosomal protein S1. J. Biol. Chem. 1994, 269, 26655–26662. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophages and Bacteria | Comments | Reference |
|---|---|---|
| Bacteriophages | ||
| φR1-RT | PCR template for phage φR1-RT genes | [14] |
| T4 | PCR template for phage T4 genes | [12] |
| fHe-Kpn01 | PCR template for phage fHe-Kpn01 genes | [10] |
| fHy-Eco03 | PCR template for phage fHy-Eco03 genes | This work |
| E. coli strains | ||
| #5509 | Human blood culture isolate | This work |
| DH10B | Cloning host | [15] |
| DH5α | Cloning host | [16] |
| DH10B/pU11L4 | Source of plasmid pU11L4 | [8] |
| Gene | Joint-Read Number % | Ratio | |
|---|---|---|---|
| Ligation Mixture | Transformant Sample | ||
| g10 | 0.86 | 0.13 | 0.15 |
| g22 | 3.10 | 0.07 | 0.02 |
| g38 | 14.84 | 26.83 | 1.81 |
| g119 | 11.70 | 19.53 | 1.67 |
| g121 | 2.79 | 15.74 | 5.63 |
| g137 | 0.48 | 13.16 | 27.2 |
| g150 | 21.38 | 3.81 | 0.18 |
| g178 | 21.74 | 10.58 | 0.49 |
| g246 | 23.11 | 10.17 | 0.44 |
| Total | 100 | 100 | |
| NGS Screening Assay Results | Plating Assay Results | ||||||
|---|---|---|---|---|---|---|---|
| Gene | % Reads in Ligation Mix | % Reads in Plasmid DNA | Ratio | SD | Gene | Fraction of g178 | SD |
| g01 | 1.31 | 3.52 | 2.69 | 0.114 | g01 | 0.680 | 0.166 |
| g03 | 1.26 | 5.33 | 4.22 | 0.546 | g03 | 1.003 | 0.015 |
| g04 | 0.89 | 2.93 | 3.30 | 0.167 | g04 | 0.672 | 0.035 |
| g05 | 3.36 | 0.01 | 0.003 | 0.001 | g05 | 0.164 | 0.003 |
| g06 | 1.46 | 3.56 | 2.43 | 0.328 | g06 | 0.684 | 0.193 |
| g08 | 1.28 | 3.85 | 3.01 | 0.196 | g08 | 0.658 | 0.130 |
| g09 | 2.80 | 4.30 | 1.53 | 0.027 | g09 | 0.827 | 0.145 |
| g10 | 1.08 | 1.63 | 1.50 | 0.334 | g10 | 0.463 | 0.186 |
| g11 | 1.07 | 2.97 | 2.78 | 0.043 | g11 | 0.901 | 0.250 |
| g13 | 2.87 | 5.71 | 1.99 | 0.202 | g13 | 1.179 | 0.428 |
| g15 | 8.64 | 5.21 | 0.60 | 0.074 | g15 | 1.104 | 0.349 |
| g17 | 7.79 | 5.49 | 0.71 | 0.014 | g17 | 0.868 | 0.224 |
| g18 | 36.90 | 41.23 | 1.12 | 0.017 | -- | -- | -- |
| g23 | 12.09 | 3.28 | 0.27 | 0.005 | g23 | 0.683 | 0.200 |
| g26 | 19.23 | 12.80 | 0.67 | 0.055 | -- | -- | -- |
| g40 | 11.47 | 3.63 | 0.32 | 0.036 | -- | -- | -- |
| g48 | 3.40 | 15.39 | 4.52 | 0.288 | g48 | 1.711 | 0.106 |
| g50 | 2.67 | 4.08 | 1.53 | 0.099 | g50 | 0.746 | 0.234 |
| g51 | 0.91 | 5.41 | 5.96 | 0.072 | g51 | 0.440 | 0.180 |
| g52 | 6.75 | 3.81 | 0.56 | 0.033 | g52 | 0.332 | 0.089 |
| g58 | 0.96 | 0.33 | 0.34 | 0.056 | -- | -- | -- |
| g59 | 2.36 | 17.27 | 7.32 | 1.827 | -- | -- | -- |
| g61 | 2.71 | 9.41 | 3.47 | 0.047 | -- | -- | -- |
| g64 | 7.91 | 2.07 | 0.26 | 0.024 | g64 | 0.332 | 0.089 |
| g65 | 5.39 | 5.40 | 1.00 | 0.049 | g65 | 0.515 | 0.140 |
| g68 | 1.31 | 1.06 | 0.81 | 0.034 | g68 | 0.451 | 0.077 |
| g69 | 8.18 | 5.62 | 0.69 | 0.023 | g69 | 0.795 | 0.237 |
| g71 | 11.59 | 10.57 | 0.91 | 0.059 | -- | -- | -- |
| g72 | 4.08 | 2.43 | 0.59 | 0.064 | -- | -- | -- |
| g73 | 10.71 | 2.34 | 0.22 | 0.019 | -- | -- | -- |
| g76 | 3.94 | 4.60 | 1.17 | 0.068 | g76 | 1.296 | 0.505 |
| g77 | 11.44 | 4.77 | 0.42 | 0.051 | g77 | 0.974 | 0.481 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasurinen, J.; Spruit, C.M.; Wicklund, A.; Pajunen, M.I.; Skurnik, M. Screening of Bacteriophage Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay. Viruses 2021, 13, 750. https://doi.org/10.3390/v13050750
Kasurinen J, Spruit CM, Wicklund A, Pajunen MI, Skurnik M. Screening of Bacteriophage Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay. Viruses. 2021; 13(5):750. https://doi.org/10.3390/v13050750
Chicago/Turabian StyleKasurinen, Jutta, Cindy M. Spruit, Anu Wicklund, Maria I. Pajunen, and Mikael Skurnik. 2021. "Screening of Bacteriophage Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay" Viruses 13, no. 5: 750. https://doi.org/10.3390/v13050750
APA StyleKasurinen, J., Spruit, C. M., Wicklund, A., Pajunen, M. I., & Skurnik, M. (2021). Screening of Bacteriophage Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay. Viruses, 13(5), 750. https://doi.org/10.3390/v13050750