
Genetic Diversity of Puumala orthohantavirus in Rodents and Human Patients in Austria, 2012–2019

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Rodent Samples
2.2. Virus Sequence Analysis
2.3. Phylogeographic Analysis
3. Results
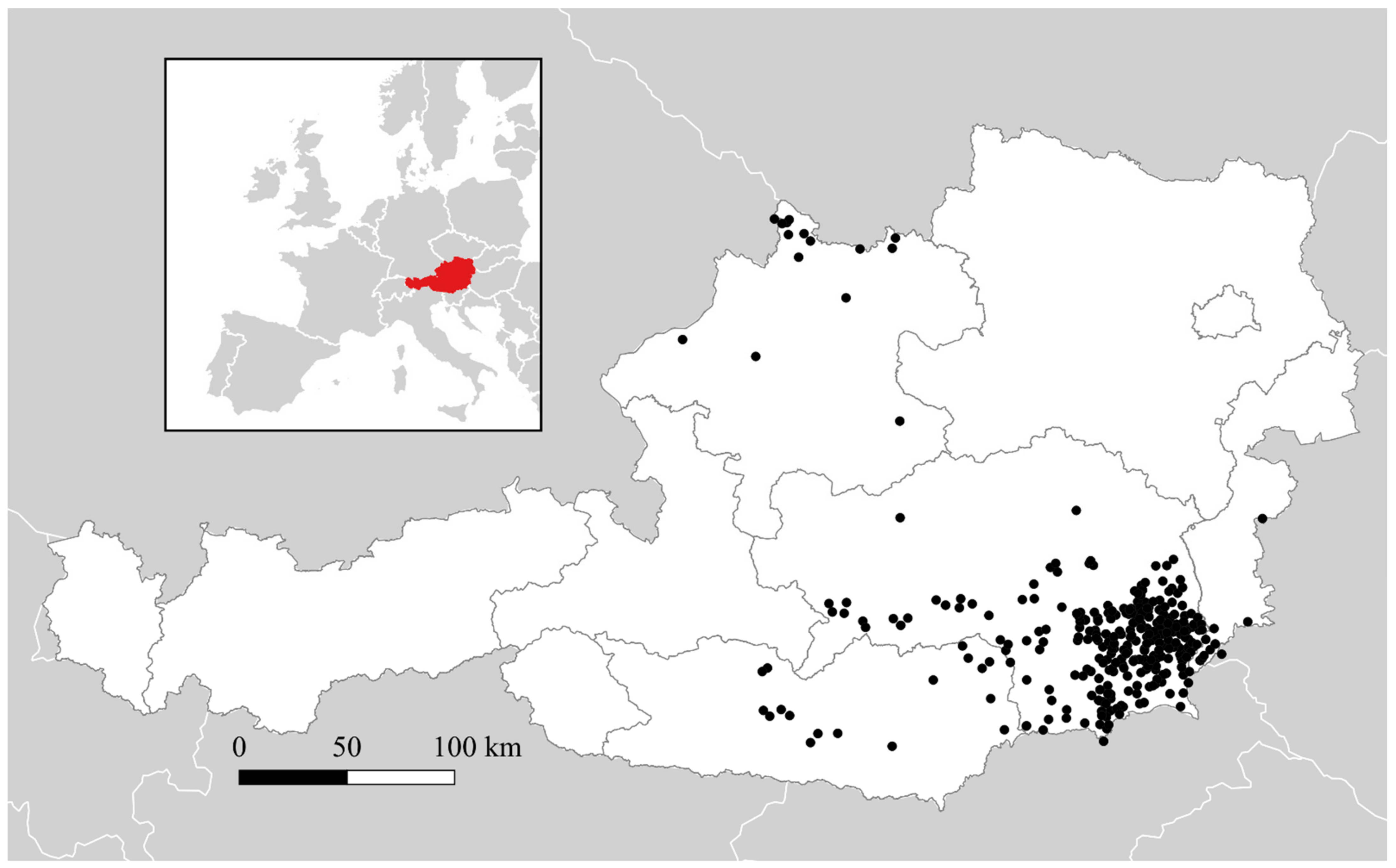
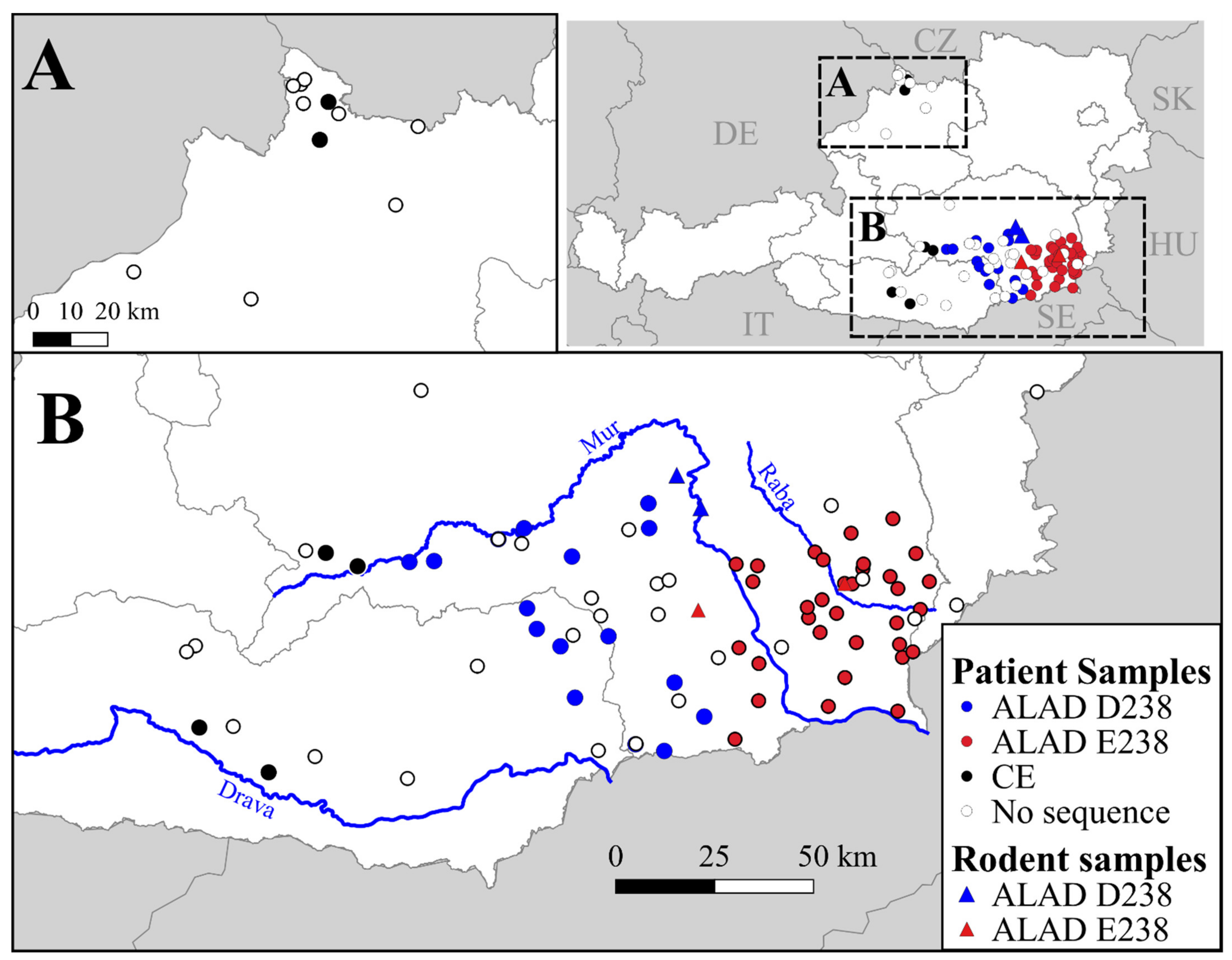
3.1. Geographic Distributrion of Puumala orthohantavirus Cases in Austria
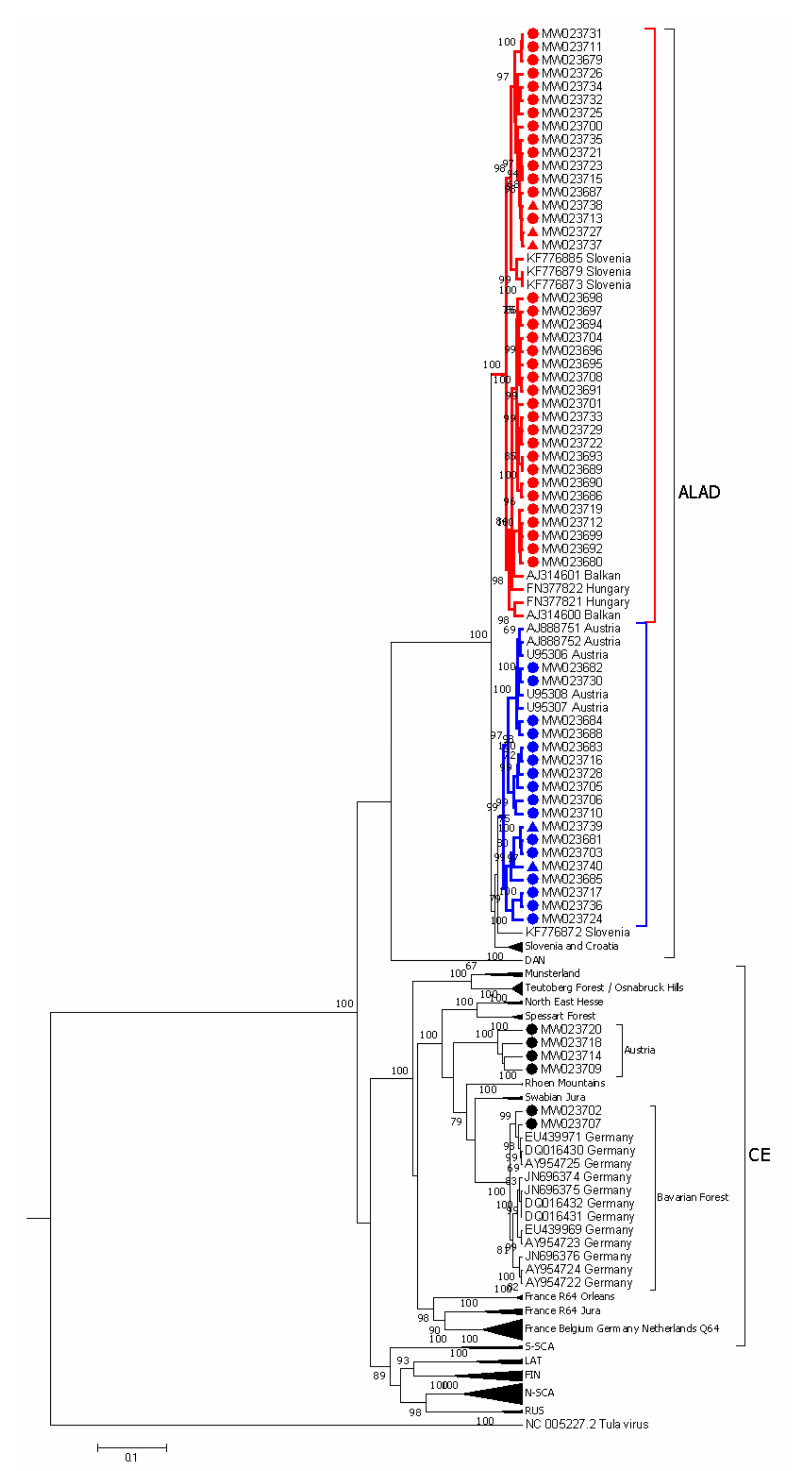
3.2. PUUV Genetic Characterization
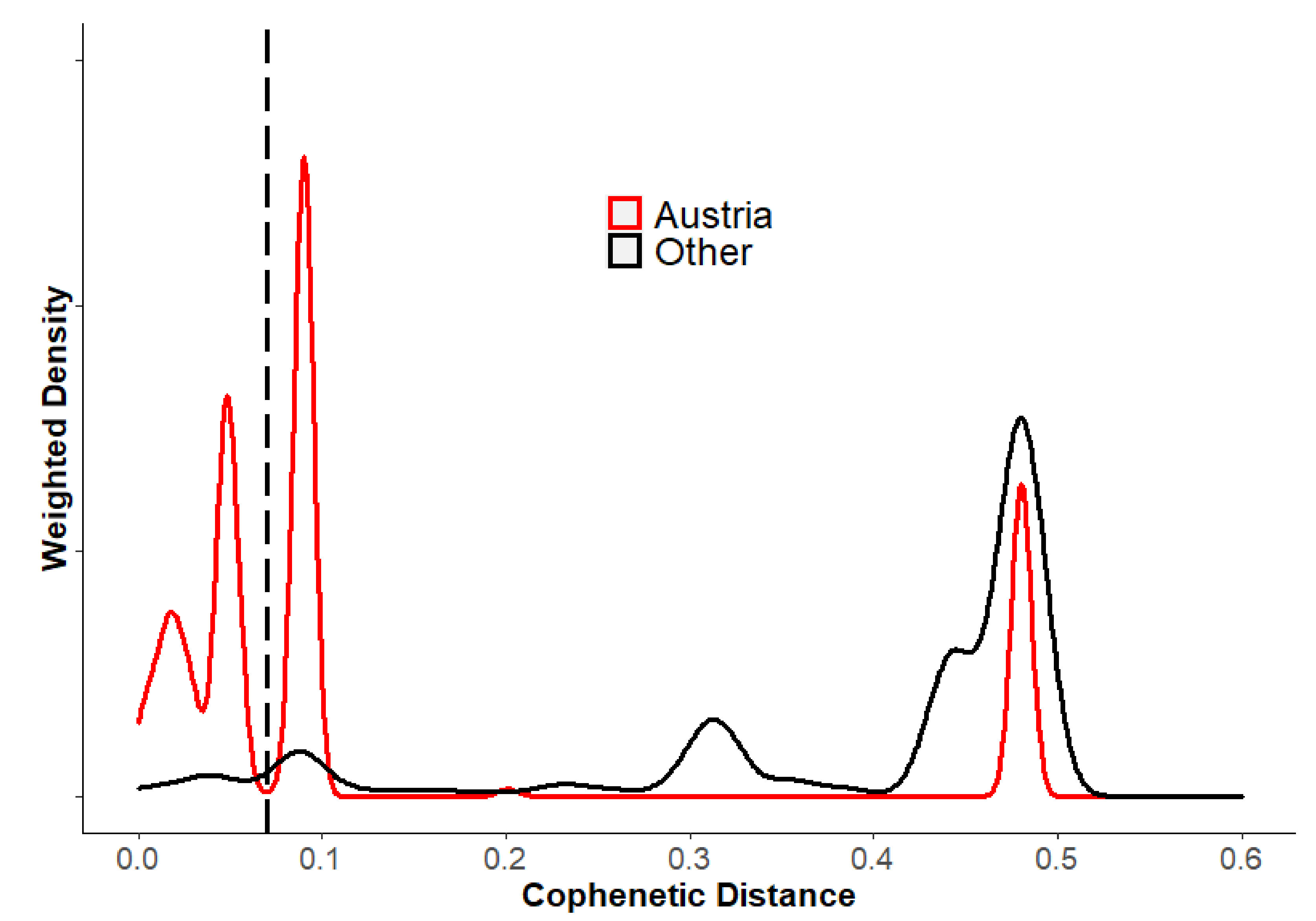
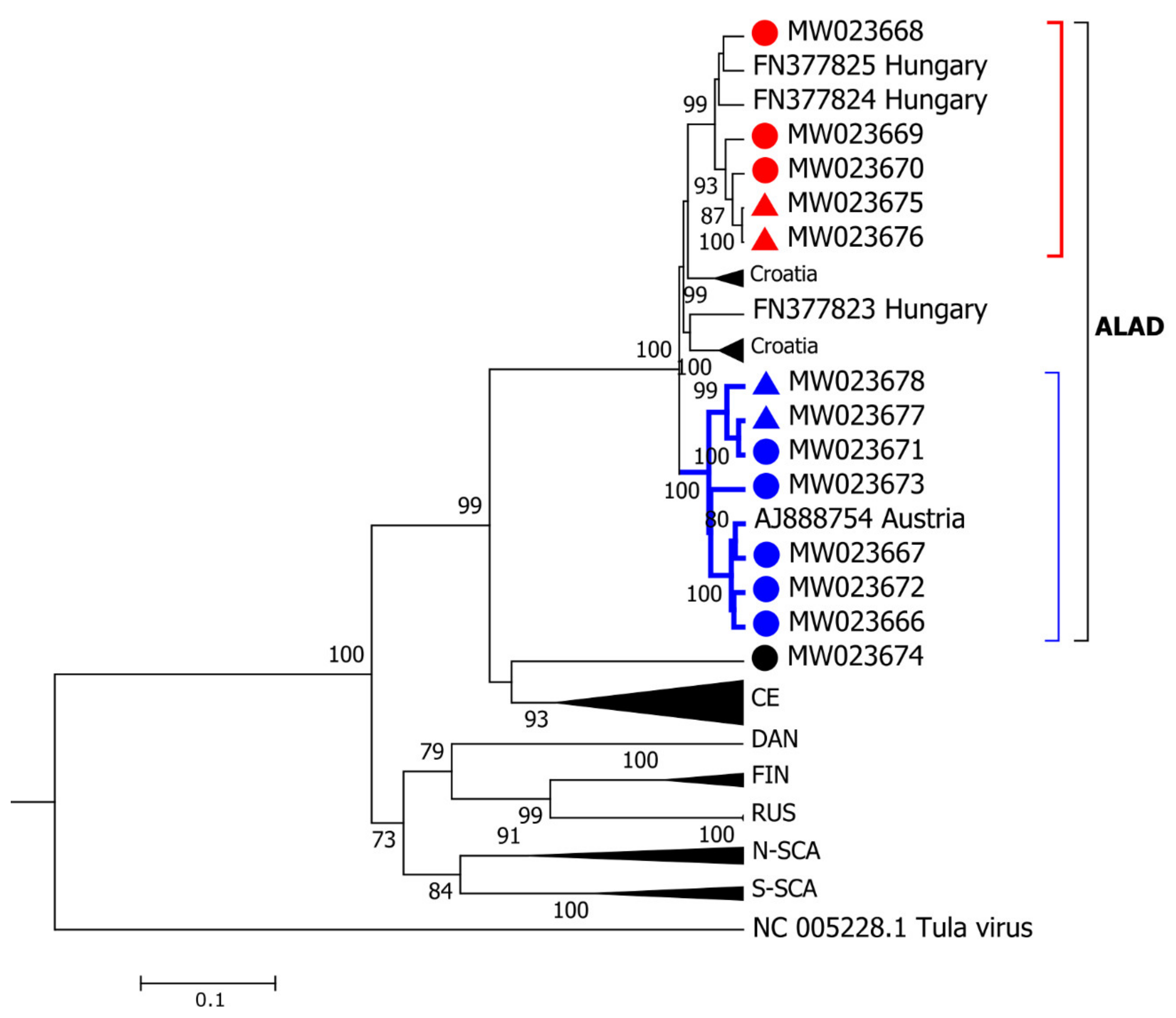
3.3. Phylogeographic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonsson, C.B.; Figueiredo, L.T.M.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Tip: Search for English results only. You can specify your search language in Preferences. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed]
- Milholland, M.T.; Castro-Arellano, I.; Suzán, G.; Garcia-Peña, G.E.; Lee, T.E.; Rohde, R.E.; Alonso Aguirre, A.; Mills, J.N. Global diversity and distribution of hantaviruses and their hosts. EcoHealth 2018, 15, 163–208. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.G.; Tsoleridis, T.; Onianwa, O.; Drake, G.; Ashpole, I.; Dobbs, P.; Edema, W.; Kumi-Ansah, F.; Bennett, M.; Tarlinton, R.E.; et al. Retrieval of the Complete Coding Sequence of the UK-Endemic Tatenale Orthohantavirus Reveals Extensive Strain Variation and Supports Its Classification as a Novel Species. Viruses 2020, 12, 454. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Hiltbrunner, M.; Drewes, S.; Ryll, R.; Wenk, M.; Spakova, A.; Petraityte-Burneikiene, R.; Heckel, G.; Ulrich, R.G. Field vole-associated Traemmersee hantavirus from Germany represents a novel hantavirus species. Virus Genes 2019, 55, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef]
- Plyusnin, A.; Hörling, J.; Kanerva, M.; Mustonen, J.; Cheng, Y.; Partanen, J.; Vapalahti, O.; Kukkonen, S.K.; Niemimaa, J.; Henttonen, H.; et al. Puumala hantavirus genome in patients with nephropathia epidemica: Correlation of PCR positivity with HLA haplotype and link to viral sequences in local rodents. J. Clin. Microbiol. 1997, 35, 1090–1096. [Google Scholar] [CrossRef]
- Voutilainen, L.; Kallio, E.R.; Niemimaa, J.; Vapalahti, O.; Henttonen, H. Temporal dynamics of Puumala hantavirus infection in cyclic populations of bank voles. Sci. Rep. 2016, 6, 21323. [Google Scholar] [CrossRef]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kuhnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef]
- Korva, M.; Knap, N.; Resman Rus, K.; Fajs, L.; Grubelnik, G.; Bremec, M.; Knapič, T.; Trilar, T.; Avšič Županc, T. Phylogeographic diversity of pathogenic and non-pathogenic hantaviruses in Slovenia. Viruses 2013, 5, 3071–3087. [Google Scholar] [CrossRef]
- Bowen, M.D.; Gelbmann, W.; Ksiazek, T.G.; Nichol, S.T.; Nowotny, N. Puumala virus and two genetic variants of tula virus are present in Austrian rodents. J. Med. Virol. 1997, 53, 174–181. [Google Scholar] [CrossRef]
- Plyusnina, A.; Aberle, S.W.; Aberle, J.H.; Plyusnin, A. Genetic analysis of Puumala hantavirus strains from Austria. Scand. j. infect. dis. 2006, 38, 512–519. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Bouckaert, R.R.; Drummond, A.J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC bioinformatics 2010, 11, 7. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing, version 4.0.3; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Hijmans, R.J. Geosphere: Spherical Trigonometry, R package, version 1.5–7; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Dietz, E.J. Permutation tests for association between two distance matrices. Systematic Zoology 1983, 32, 21–26. [Google Scholar] [CrossRef]
- Mondaca-Fernández, E.; Murtaugh, M.P.; Morrison, R.B. Association between genetic sequence homology of Porcine reproductive and respiratory syndrome virus and geographic distance between pig sites. Can. J. Vet. Res. 2006, 70, 237–239. [Google Scholar]
- Real, L.A.; Henderson, J.C.; Biek, R.; Snaman, J.; Jack, T.L.; Childs, J.E.; Stahl, E.; Waller, L.; Tinline, R.; Nadin-Davis, S. Unifying the spatial population dynamics and molecular evolution of epidemic rabies virus. Proc. Natl. Acad. Sci. USA 2005, 102, 12107–12111. [Google Scholar] [CrossRef] [PubMed]
- Binder, F.; Ryll, R.; Drewes, S.; Jagdmann, S.; Reil, D.; Hiltbrunner, M.; Rosenfeld, U.M.; Imholt, C.; Jacob, J.; Heckel, G.; et al. Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment. Pathogens 2020, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. Biol. Sci. 2017, 284, 20170857. [Google Scholar] [CrossRef]
- Hiltbrunner, M.; Heckel, G. Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples. Viruses 2020, 12, 749. [Google Scholar] [CrossRef] [PubMed]
- Faber, M.; Krüger, D.H.; Auste, B.; Stark, K.; Hofmann, J.; Weiss, S. Molecular and epidemiological characteristics of human Puumala and Dobrava-Belgrade hantavirus infections, Germany, 2001 to 2017. Eurosurveillance 2019, 24, 1800675. [Google Scholar] [CrossRef] [PubMed]
- Essbauer, S.; Schmidt, J.; Conraths, F.J.; Friedrich, R.; Koch, J.; Hautmann, W.; Pfeffer, M.; Wolfel, R.; Finke, J.; Dobler, G.; et al. A new Puumala hantavirus subtype in rodents associated with an outbreak of Nephropathia epidemica in South-East Germany in 2004. Epidemiol. Infect. 2006, 134, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Wölfel, R.; Ullrich, K.; Yoshimatsu, K.; Blumhardt, J.; Römer, I.; Esser, J.; Schmidt-Chanasit, J.; Groschup, M.H.; Dobler, G.; et al. Seroepidemiological study in a Puumala virus outbreak area in South-East Germany. Med. Microbiol. Immunol. 2009, 198, 83–91. [Google Scholar] [CrossRef]
- Filippone, C.; Castel, G.; Murri, S.; Ermonval, M.; Korva, M.; Avsic-Zupanc, T.; Sironen, T.; Vapalahati, O.; McElhinney, L.M.; Ulrich, R.G.; et al. Revisiting the genetic diversity of emerging hantaviruses circulating in Europe using a pan-viral resequencing microarray. Sci. Rep. 2019, 9, 12404. [Google Scholar] [CrossRef]
- Aberle, S.W.; Lehner, P.; Ecker, M.; Aberle, J.H.; Arneitz, K.; Khanakah, G.; Radda, A.; Radda, I.; Popow-Kraupp, T.; Kunz, C.; et al. Nephropathia epidemica and Puumala virus in Austria. Eur. J. Clin. Microbiol. Infect. Dis. 1999, 18, 467–472. [Google Scholar] [CrossRef]
- Plyusnina, A.; Ferenczi, E.; Racz, G.R.; Nemirov, K.; Lundkvist, A.; Vaheri, A.; Vapalahti, O.; Plyusnin, A. Co-circulation of three pathogenic hantaviruses: Puumala, Dobrava, and Saaremaa in Hungary. J. Med. Virol. 2009, 81, 2045–2052. [Google Scholar] [CrossRef]
- Drewes, S.; Ali, H.S.; Saxenhofer, M.; Rosenfeld, U.M.; Binder, F.; Cuypers, F.; Schlegel, M.; Rohrs, S.; Heckel, G.; Ulrich, R.G. Host-Associated Absence of Human Puumala Virus Infections in Northern and Eastern Germany. Emerg. Infect. Dis. 2017, 23, 83–86. [Google Scholar] [CrossRef]
- Ali, H.S.; Drewes, S.; Weber de Melo, V.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef]
- Mertens, M.; Kindler, E.; Emmerich, P.; Esser, J.; Wagner-Wiening, C.; Wolfel, R.; Petraityte-Burneikiene, R.; Schmidt-Chanasit, J.; Zvirbliene, A.; Groschup, M.H.; et al. Phylogenetic analysis of Puumala virus subtype Bavaria, characterization and diagnostic use of its recombinant nucleocapsid protein. Virus Genes 2011, 43, 177–191. [Google Scholar] [CrossRef]
- Markova, S.; Hornikova, M.; Lanier, H.C.; Henttonen, H.; Searle, J.B.; Weider, L.J.; Kotlik, P. High genomic diversity in the bank vole at the northern apex of a range expansion: The role of multiple colonizations and end-glacial refugia. Mol. Ecol. 2020, 29, 1730–1744. [Google Scholar] [CrossRef]
- Szabo, R.; Radosa, L.; Lickova, M.; Slavikova, M.; Heroldova, M.; Stanko, M.; Pejcoch, M.; Osterberg, A.; Laenen, L.; Schex, S.; et al. Phylogenetic analysis of Puumala virus strains from Central Europe highlights the need for a full-genome perspective on hantavirus evolution. Virus Genes 2017, 53, 913–917. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camp, J.V.; Schmon, E.; Krause, R.; Sixl, W.; Schmid, D.; Aberle, S.W. Genetic Diversity of Puumala orthohantavirus in Rodents and Human Patients in Austria, 2012–2019. Viruses 2021, 13, 640. https://doi.org/10.3390/v13040640
Camp JV, Schmon E, Krause R, Sixl W, Schmid D, Aberle SW. Genetic Diversity of Puumala orthohantavirus in Rodents and Human Patients in Austria, 2012–2019. Viruses. 2021; 13(4):640. https://doi.org/10.3390/v13040640
Chicago/Turabian StyleCamp, Jeremy V., Eva Schmon, Robert Krause, Wolfdieter Sixl, Daniela Schmid, and Stephan W. Aberle. 2021. "Genetic Diversity of Puumala orthohantavirus in Rodents and Human Patients in Austria, 2012–2019" Viruses 13, no. 4: 640. https://doi.org/10.3390/v13040640
APA StyleCamp, J. V., Schmon, E., Krause, R., Sixl, W., Schmid, D., & Aberle, S. W. (2021). Genetic Diversity of Puumala orthohantavirus in Rodents and Human Patients in Austria, 2012–2019. Viruses, 13(4), 640. https://doi.org/10.3390/v13040640